




Abstract

Background: Systemic lupus erythematosus (SLE) is a complex autoimmune disease that severely impacts patient quality of life. Current treatments primarily manage symptoms rather than cure the disease, emphasizing the need for a deeper understanding of its pathogenesis and the discovery of novel therapeutic targets. Circulating proteins are thought to play a critical role in SLE risk, but their causal relationships remain underexplored.
Methods: This study used pQTL and GWAS data to perform a two-sample Mendelian randomization (MR) analysis to investigate the genetic causal relationships between circulating proteins and SLE. We identified proteins potentially associated with SLE risk and further analyzed their roles in immune regulation and inflammation using Protein-Protein Interaction (PPI) networks. Colocalization analysis was conducted to validate the associations of key proteins with SLE.
Results: Our analysis identified 82 plasma proteins potentially causally linked to SLE risk. Colocalization analysis confirmed the association of proteins such as TNFAIP3, PDHX, and CTSF with SLE, underscoring their critical role in disease pathogenesis. Additionally, Protein-Protein Interaction (PPI) network analysis revealed that these proteins are involved in immune modulation and inflammatory pathways, further supporting their relevance as therapeutic targets.
Conclusion: This study identifies 82 plasma proteins that may play a causal role in SLE, with TNFAIP3, PDHX, and CTSF emerging as promising therapeutic targets. These findings provide a foundation for future research aimed at developing precision therapies for SLE.
Keywords: Systemic Lupus Erythematosus; Mendelian Randomization; Circulating Proteins; Colocalization Analysis; Protein-Protein Interaction Network
Introduction
Systemic lupus erythematosus (SLE) is a chronic, multi-system autoimmune disorder that profoundly affects the quality of life and imposes significant socio-economic burdens[1, 2]. The conventional treatment regimen for SLE includes nonsteroidal anti-inflammatory drugs (NSAIDs), antimalarials, corticosteroids, and immunosuppressants[3]. . Recent advancements have introduced biologic agents, offering new therapeutic avenues for SLE management. For instance, belimumab, approved by the U.S. Food and Drug Administration (FDA), targets the B-lymphocyte stimulator (BLyS) to curtail autoantibody production[4]. Despite the diversity of treatment options, therapeutic outcomes for SLE vary considerably among individuals, and most strategies prioritize symptom management over disease eradication[5] .Moreover, long-term use of existing medications can lead to serious adverse effects, including infections, osteoporosis, and retinal disorders[5].
Proteins play a crucial role in immune regulation and inflammatory responses, both of which are closely associated with the pathophysiological processes of SLE[6]. For example, complement activation is one of the key pathological mechanisms leading to tissue inflammation and damage in systemic lupus erythematosus (SLE)[7]. Furthermore, proteins are primary targets for pharmacological interventions[8]. In two Phase III trials, anifrolumab, which blocks the type I interferon receptor 1 (IFNAR1), received approval for treating SLE[9]. The circulating proteome, comprising proteins released both actively and passively into the bloods from various tissues and cells[10], has been confirmed its correlation with SLE. Yong Dai's team utilized data-dependent acquisition (DDA) and data-independent acquisition (DIA) proteomics techniques to identify three proteins as potential biomarkers for diagnosing SLE[11]. Liu et al. applied TMT-labeled quantitative proteomics alongside enzyme-linked immunosorbent assays (ELISA) to demonstrate notable differences in the serum levels of SAA1 and CD248 between SLE patients and healthy controls[12]. Nonetheless, current research is constrained by small sample sizes, a limited protein range, and potential confounding factors. Conducting randomized controlled trials to investigate the potential causal relationships between numerous proteins and SLE is also challenging.
Mendelian Randomization (MR) employs genetic variations as instrumental variables (IVs) to establish causal relationships between exposures and outcomes, effectively mitigating confounding factors and preventing reverse causality[13, 14]. In this study, we conducted a two-sample Mendelian randomization analysis using pQTL data derived from extensive proteomics studies and genome-wide association study (GWAS) data for SLE to explore the genetic causal relationship between these elements. Additionally, we constructed a protein-protein interaction (PPI) network and performed colocalization analysis for proteins statistically significant in the MR analysis. This effort aims to identify potential therapeutic targets and lay the foundation for future clinical applications (Figure 1).
Figure 1. The flow chart of the overall study design Mendelian Randomization (MR) explores genetic causal links between plasma proteins and systemic lupus erythematosus (SLE). Using data from 4,775 plasma proteins (Fenland study) and GWAS of SLE (FinnGen: 1,083 cases, 306,504 controls), cis-pQTL variables meeting stringent thresholds (R² < 0.001, P < 5 × 10⁻⁸, F-statistics > 10) serve as instruments. MR identifies 82 proteins with causal effects on SLE risk, with further analysis performed using protein-protein interaction networks and colocalization studies.

Methods
Source of Exposure Data
We obtained single nucleotide polymorphism (SNP) data associated with plasma
protein levels from the Fenland study.This research carried out a genome-wide proteomics association study involving
10,708 participants of European ancestry, assessing 4,775 plasma proteins utilizing the SomaScan v4 assay
(http://www.omicscience.org/apps/pgwas)[15].
Source of Outcome Data
The outcome of interest, systemic lupus erythematosus (SLE), was studied using
genome-wide association study (GWAS) data obtained from the Finnish Genetic Study (FinnGen). This study extracted over
500,000 samples from the Finnish biobank, integrating longitudinal phenotypic data and digital health records from the
national health registry[16]. We accessed the publicly available FinnGen R10 dataset
(https://r10.finngen.fi/), which includes data on 1,083 SLE cases and 306,504 controls[16].
Study Design
In this research, we performed a comprehensive two-sample Mendelian Randomization (MR)
analysis to evaluate the causal relationship between circulating proteins and systemic lupus erythematosus (SLE). To
ensure the validity of the results, the MR analysis mandated that the selected instrumental variables (IVs) satisfy
three critical criteria: (1) the IVs must demonstrate a significant direct association with the exposure variable
(circulating proteins); (2) the IVs must be independent of any confounders that could affect the exposure-outcome
relationship; (3) the influence of the IVs on the outcome must be mediated exclusively through the exposure, with no
alternative causal pathways[17].
Selection of Instrumental Variables
Informed by the three assumptions outlined previously and recent research findings,stringent criteria for SNP
selection: (1) SNPs must be significantly associated with circulating proteins, adhering to a stringent significance
threshold (P-value < 5 × 10-8)[18]; (2) To ensure the independence of the selected IVs and mitigate
the effects of linkage disequilibrium (LD), SNP clustering methods were utilized (r² = 0.001, kb = 10,000)[19]; (3) IVs with an F-statistic less than 10, generally considered weak, were excluded to avoid instability and bias
in effect estimation[20]; (4) SNPs strongly associated with the outcome variable (P-value < 5 ×
10-8) were also excluded to prevent direct causal interference with the outcome[21]; (5)
Cis-protein quantitative trait loci (cis-pQTLs) spanning a gene range of ±1Mb were selected. Located at or near the
gene encoding the target proteins, cis-QTLs are favored for their substantial contribution to explaining protein
expression, compared to variants in other genomic regions[22].
Mendelian Randomization Analysis and Sensitivity Analysis
In this study, we conducted two-sample MR
analysis using R software (version 4.3.3) and the "TwoSampleMR" package, employing various statistical methods to
assess the potential causal relationship between circulating proteins and the risk of systemic lupus erythematosus
(SLE). We employed the inverse variance weighted (IVW) method as the primary analytical tool when two or more
instrumental variables (IVs) were available. Additionally, when the number of IVs was three or more, the weighted
median (WM) and Mendelian randomization-Egger (MR-Egger) methods were also implemented. For proteins represented by a
single IV, the Wald ratio method was applied to estimate the change in the log odds ratio of SLE risk per one standard
deviation (SD) increase in protein levels[23].
Additionally, we performed sensitivity analysis using Cochran's Q test, MR-Egger, and MR-PRESSO methods. The Cochran's Q statistic assessed heterogeneity among the selected instrumental variables, with a P-value < 0.05 denoting significant heterogeneity. A significant intercept in the MR-Egger method indicated the potential pleiotropy, which was determined using a P-value < 0.05[24]. The MR-PRESSO method, utilized via the "MR-PRESSO" package, aimed to identify and remove SNP outliers with horizontal pleiotropy. However, when the number of SNPs is small, it is insufficient for effective heterogeneity and pleiotropy analysis[25]. Lastly, the MR-Steiger test was applied to evaluate directional causality by comparing the proportion of variance explained by the instrumental variables in relation to both the exposure and outcome variables, thus assessing the suitability of the instrumental variables[26].
pQTL - GWAS Colocalization Analysis
To determine whether protein expression levels and SLE risk share
causal variants within the same genomic region, we performed a colocalization analysis using the "coloc" R package
with default prior probabilities[27]. Bayesian methods were applied to each cis-gene locus of the
proteins to evaluate five mutually exclusive hypotheses: (1) No significant association with either trait (H0); (2)
Associated only with protein levels (H1); (3) Associated only with SLE risk (H2); (4) Association with both traits,
driven by distinct causal variants (H3); (5) Both traits driven by the same causal variant (H4). In our analysis,
co-localization was deemed supported when the posterior probability of sharing pathogenic variants (PP.H4) exceeded
0.6[28].
Protein-Protein Interaction Network
To enhance our understanding of the interactions between proteins, we
constructed a protein-protein interaction (PPI) network. Using the STRING database (https://cn.string-db.org/), we
developed the PPI network with a minimum interaction threshold of 0.4, maintaining other parameters at their default
settings[29].
Results
Circulating proteins and SLE risk mendelian analysis results
In this study, due to the stringent selection
criteria for instrumental variables, only 1445 circulating proteins were included in the analysis (Supplementary Table 1). Mendelian randomization (MR) analysis revealed that 82 plasma proteins were potentially causally associated with
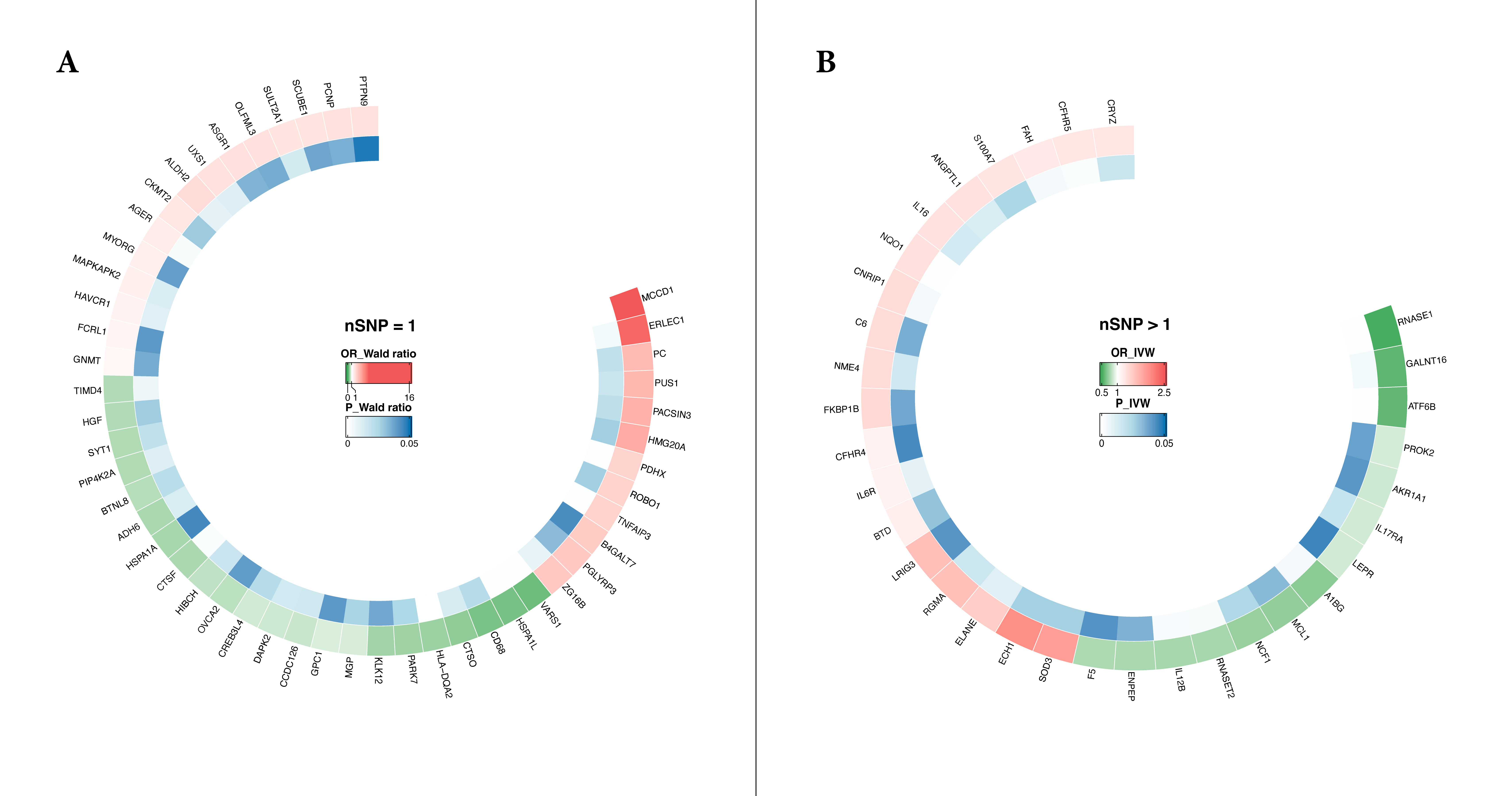
SLE risk (P < 0.05) as shown in Figure 2. Specifically, high expression of 46 proteins and low
expression of 36 proteins are positively correlated with an increased risk of systemic lupus erythematosus (SLE).
Additionally, for proteins with at least three SNPs (nSNP ≥ 3), 13 proteins were further validated by the Weighted
Median (WM) method as being associated with SLE risk (P < 0.05), while no specific associations were identified by the
MR Egger method (Supplementary Table 2).
Figure 2. Mendelian Randomization Analysis of 82 Circulating Proteins Associated with SLE Risk (P < 0.05) The circular heatmap illustrates the results of Mendelian Randomization (MR) analysis for 82 circulating proteins with potential causal relationships to systemic lupus erythematosus (SLE) (p < 0.05). Panel A presents the analysis using the Wald ratio as the primary method when the number of SNPs (nSNP) equals 1. Panel B shows the results when the number of SNPs (nSNP) exceeds 1, with inverse variance weighting (IVW) as the primary analysis method.
The minimum F-statistic for the selected instrumental variables (IVs) for each protein was 23.926, confirming their strength as robust instruments. The p-values of the Steiger test ranged from 0 to 9.01E-06, affirming that the directionality of the instrumental variables is consistent with the fundamental assumptions of the MR analysis. Additionally, the MR-Egger intercept tests for each protein indicated no evidence of horizontal pleiotropy (P > 0.05), with further details available in Supplementary Table 2.
Colocalization Analysis
Bayesian co-localization analysis was conducted on 82 circulating proteins that
showed statistically significant associations, encompassing both upstream and downstream regions (detailed results are
presented in Supplementary Table 3). The analysis revealed co-localization between systemic lupus erythematosus
(SLE) and several proteins, namely PDHX (PP.H4 = 0.67), CTSF (PP.H4 = 0.64), and TNFAIP3 (PP.H4 = 0.66), suggesting
that these proteins may share common genetic variants with SLE (Figure 3).
Figure 3. Colocalization Analysis of TNFAIP3, PDHX, and CTSF with Systemic Lupus Erythematosus Risk. Colocalization analysis identifies shared genetic variants influencing plasma protein levels of TNFAIP3 (A), PDHX (B), and CTSF (C) and systemic lupus erythematosus (SLE) risk. The top panels show pQTLs for plasma protein levels, and the bottom panels display genetic associations with SLE. Key variants, including rs59693083 (TNFAIP3), rs12289762 (PDHX), and rs4920540 (CTSF), exhibit significant colocalization, indicating shared causal variants for protein expression and SLE.

PPI networks
A total of 82 proteins (P < 0.05) were entered into the STRING database to construct a
protein network. Given the study's threshold for minimum interaction strength of 0.4, only 50 imported proteins
successfully formed a network with other supplementary proteins, collectively comprising 82 nodes and 54 edges (Figure 4). Subsequent network functional enrichment analysis (Supplementary Figure 1) identified several proteins significantly involved in biological processes such as inflammatory response and immune
system regulation, with PFDR values of 1.1e-04 and 4.8e-04, respectively. These processes are intimately connected to
the pathophysiology of SLE.[30, 31].
Figure 4. Protein-Protein Interaction (PPI) Network of Proteins Associated with SLE Risk The PPI network shows interactions among plasma proteins linked to SLE risk, with nodes representing proteins and edges indicating interactions from the STRING database. Key hubs like TNFAIP3, PDHX, and IL6R highlight functionally related clusters, suggesting pathways involved in SLE pathogenesis.

Discussion
This proteomics Mendelian Randomization study investigated the causal relationships between 4775 plasma proteins and the risk of systemic lupus erythematosus (SLE). Employing stringent selection criteria, we identified 82 plasma proteins significantly associated with SLE risk. Notably, proteins such as TNFAIP3, PDHX, and CTSF appear to share the same causal variants as the disease. We further elucidated the intricate biological interactions among these proteins using the Protein-Protein Interaction (PPI) network.
TNFAIP3 (TNF Alpha Induced Protein 3) is an ubiquitin-editing enzyme extensively documented to act as an endogenous negative feedback regulator of inflammatory responses by inhibiting NF-κB signaling pathway activity[32, 33]. However, TNFAIP3 also promotes the phosphorylation of receptor-interacting protein 3 (RIP3) through deubiquitination[34], thereby activating the NLRP3 inflammasome pathway, which contributing to the development of lupus nephritis[35].Additionally, various GWAS studies across different populations have demonstrated that single nucleotide polymorphisms (SNPs) at the TNFAIP3 locus are associated with susceptibility to SLE[36]. For instance, the rs2230926 SNP alters the amino acid sequence of the A20 protein (from phenylalanine to cysteine), diminishing its capacity to inhibit TNF-induced NF-κB activation. This alteration compromises the inflammatory control in individuals carrying this risk allele, thereby heightening their susceptibility to SLE[37, 38]. Additionally, cohort studies have associated the TNFAIP3 rs5029939 genetic polymorphism with SLE susceptibility and potential impacts on its clinical phenotype[38]. Notably, this study identifies for the first time that rs59693083 (located in the TNFAIP3 promoter[39]) escalates the risk of SLE, offering new insights into the pathogenesis and clinical treatment of the disease.
PDHX, a crucial component of the pyruvate dehydrogenase complex (PDC), primarily facilitates the conversion of pyruvate into acetyl-CoA, bridging glycolysis to the Krebs cycle[40]. Research indicates that the 11p13 locus, situated between PDHX and CD44, is linked to genetic susceptibility to SLE. This region contains multiple regulatory sites, which potentially affect the expression and function of both PDHX and CD44, consequently influencing immune regulation and inflammatory responses in SLE[41]. This aligns with our MR results. Intriguingly, high PDHX expression has also been associated with diminished immune cell infiltration in the tumor microenvironment[42], suggesting that PDHX's role in SLE pathogenesis might be more complex than previously understood. These insights necessitate further basic and clinical studies to elucidate the underlying mechanisms.
CTSF (Cathepsin F) is a lysosomal cysteine protease integral to various physiological and pathological processes, including immune responses and antigen processing[43]. However, its impact on the immune system is subject to ongoing debate. Evidence indicates that inhibiting CTSF activity may block MHC class II processing in macrophages, reducing antigen presentation by MHC class II, and potentially ameliorating diseases associated with inappropriate or excessive immune responses[44]. However, In clear cell renal carcinoma (ccRCC) studies, CTSF expression inversely correlated with both the infiltration of diverse immune cells and the expression of MHC molecules such as TAP1 and TAP2[45]. Our MR results indicate a negative correlation between CTSF and SLE risk. Additionally, some studies provide indirect evidence. For example, cysteine cathepsins (Cts) have been implicated in the hydrolysis of the extracellular matrix (ECM), involving the processing of cytokines, chemokines, and cell adhesion molecules, which are pivotal in inflammatory responses[46].. Cts may mitigate inflammation triggered by cellular debris by efficiently degrading damaged organelles and proteins. Although the link between CTSF and SLE risk has been less extensively explored, the causal relationship identified in this study invites further investigation into its specific role and mechanisms in SLE and immune system regulation, potentially unveiling new therapeutic targets.
This study employed a large-scale dataset for Mendelian Randomization analysis to enhance result reliability. In the initial phases, we selected cis-pQTLs for inclusion due to their typical proximity to or within genes encoding proteins, thus directly regulating protein expression by affecting transcription, translation, degradation, stability, or activity. Additionally, our team utilized various statistical methods such as the Wald ratio, Inverse Variance Weighted (IVW), Weighted Median (WM), and Mendelian Randomization-Egger (MR-Egger) to bolster the robustness of the MR analysis. Finally, we performed protein-protein interaction (PPI) network analysis to validate the links between protein-related biological processes and the disease's pathological processes.
Despite the strengths of our study, it has several limitations. Firstly, it relies on population data from individuals of European ancestry, thereby constraining the generalizability of the findings to other ancestral groups. Secondly, while the Fenland study included a broad range of circulating proteins, our stringent selection criteria for instrumental variables might have excluded other relevant target proteins. Moreover, protein expression and function are subject to influences from various factors, such as environmental interactions and epigenetic modifications, which were not comprehensively accounted for in our analysis. These complex influences extend beyond genetic variations and were not thoroughly addressed in this study.
Conclusion
Overall, this study offers a comprehensive assessment of the causal relationships between circulating proteins and systemic lupus erythematosus (SLE), further confirming their critical roles in the initiation and progression of the disease. Notably, proteins such as TNFAIP3, PDHX, and CTSF are identified as likely candidates for new therapeutic targets. However, additional research is essential to unravel the complex mechanisms linking these candidate proteins with SLE risk, a crucial step in validating their potential clinical relevance.
Abbreviations
B-lymphocyte stimulator: BLyS; cysteine cathepsins: Cts; Cis-protein Quantitative Trait Loci: cis-pQTLs; data-dependent acquisition: DDA; data-independent acquisition: DIA; Extracellu-lar Matrix: ECM; enzyme-linked immunosorbent assays: ELISA; Food and Drug Administration: FDA; Genome - Wide Associa-tion Study: GWAS; I interferon receptor 1: IFNAR1; Instrumen-tal Variables: IVs; Inverse Variance Weighted: IVW; Linkage Disequilibrium: LD; Mendelian Randomization: MR; Mendelian Randomization-Egger: MR-Egger; protein Quantitative Trait Loci: pQTL; Protein-Protein Interaction: PPI; Receptor-Interact-ing Protein 3: RIP3; Single Nucleotide Polymorphisms: SNPs; Systemic Lupus Erythematosus: SLE; Trans-protein Quantita-tive Trait Loci: trans-pQTLs; Weighted Median: WM.
Supplementary Materials
Declarations
Author contributions
Xinzhen Zhao: Writing – review & editing, original draft, Conceptualization, Visualization, Formal analysis. Yinying Chai: Writing – review & editing, Validation, Supervision, Data curation, Formal analysis. Qianran Hong: Writing – review & editing, Writing – original draft, Validation, Funding acquisition, Formal analysis. Yuxuan Song: Writing – review & editing, Validation, Funding acquisition, Data curation,. Yibo He: Writing – review & editing, Conceptualization, Supervision.
Acknowledgements
Not Applicable.
Funding information
This work was supported by Innovation Fund for Qutstanding Doctoral Candidates of Peking University Health Science Center (BMU2024BSS001).
Ethics approval and consent to participate
Not Applicable.
Competing Interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
The data that supports the findings of this study are available in the supplementary material of this article
References
 Figures
Figures References
References Peer
Peer Information
InformationFigure 1. The flow chart of the overall study design Mendelian Randomization (MR) explores genetic causal links between plasma proteins and systemic lupus erythematosus (SLE). Using data from 4,775 plasma proteins (Fenland study) and GWAS of SLE (FinnGen: 1,083 cases, 306,504 controls), cis-pQTL variables meeting stringent thresholds (R² < 0.001, P < 5 × 10⁻⁸, F-statistics > 10) serve as instruments. MR identifies 82 proteins with causal effects on SLE risk, with further analysis performed using protein-protein interaction networks and colocalization studies.
Figure 2. Mendelian Randomization Analysis of 82 Circulating Proteins Associated with SLE Risk (P < 0.05) The circular heatmap illustrates the results of Mendelian Randomization (MR) analysis for 82 circulating proteins with potential causal relationships to systemic lupus erythematosus (SLE) (p < 0.05). Panel A presents the analysis using the Wald ratio as the primary method when the number of SNPs (nSNP) equals 1. Panel B shows the results when the number of SNPs (nSNP) exceeds 1, with inverse variance weighting (IVW) as the primary analysis method.
Figure 3. Colocalization Analysis of TNFAIP3, PDHX, and CTSF with Systemic Lupus Erythematosus Risk. Colocalization analysis identifies shared genetic variants influencing plasma protein levels of TNFAIP3 (A), PDHX (B), and CTSF (C) and systemic lupus erythematosus (SLE) risk. The top panels show pQTLs for plasma protein levels, and the bottom panels display genetic associations with SLE. Key variants, including rs59693083 (TNFAIP3), rs12289762 (PDHX), and rs4920540 (CTSF), exhibit significant colocalization, indicating shared causal variants for protein expression and SLE.
Figure 4. Protein-Protein Interaction (PPI) Network of Proteins Associated with SLE Risk The PPI network shows interactions among plasma proteins linked to SLE risk, with nodes representing proteins and edges indicating interactions from the STRING database. Key hubs like TNFAIP3, PDHX, and IL6R highlight functionally related clusters, suggesting pathways involved in SLE pathogenesis.
Peer-review Terminology
Identity transparency: Single anonymized
Reviewer interacts with: Editor
Review information published:
Review reports
Reviewer identities if reviewer opts in
Author/reviewer communication
Details
© 2025 The Author(s). Life Conflux published by Life Conflux Press Limited on behalf of Conflux Science.
This is an open access article under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Publication History
Received 2025-06-17
Accepted 2025-07-14
Published 2025-08-15