




Abstract
The cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway, a central hub of the innate immune system, is a key me-diator of immune surveillance against abnormal cytoplasmic dsDNA: cGAS recognizes such dsDNA to synthesize 2'3'-cGAMP, which activates STING and downstream signaling to drive IFN-I and proinflammatory cytokine expression for the maintenance of homeostasis. This mechanism enables the pathway to exert multidimensional roles in physiology and pathology. Its activity is fine-tuned by post-translational modifications and non-coding RNAs. Given its critical role in linking innate immunity to disease progression, it has become a promising therapeutic target. This review summarizes the pathway’s regulatory mechanisms and pathological implications, detailing its roles in immune activation, disease dysreg-ulation, and therapeutic development. It also addresses existing challenges and proposes future directions, aiming to provide new insights for precision therapy against cGAS-STING-associated diseases.
Keywords: cGAS; STING; Inflammation; Tumor Immunity; Diseases therapy.
Introduction
The innate immune system is the first line of defense against external pathogens, playing a crucial role in immune responses. As a major component of the immune system, the innate immune system not only provides timely defense responses at the onset of infection but also triggers a series of immune reactions by recognizing exogenous pathogens and endogenous damage signals, thereby maintaining bodily homeostasis [1]. Unlike adaptive immunity, the innate immune system does not rely on prior immunological memory. Instead, it directly recognizes pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) through a wide array of pattern recognition receptors (PRRs), which rapidly activate inflammatory responses, initiate antiviral mechanisms, and regulate immune balance [2, 3].
In innate immune responses, the cGAS-STING pathway has become a research hotspot in recent years and has been shown to play a critical role in various immune responses [4]. The cGAS-STING signaling pathway is a central immune signaling pathway in both the innate immune system and intracellular signaling. It serves as a key “DNA-sensing” pathway, deeply involved in the host’s immune response to exogenous pathogens (such as viruses and bacteria) and responses to endogenous damage [5–7]. As a DNA sensor in this pathway, cGAS recognizes double-stranded DNA (dsDNA) that is abnormally present in the cytoplasm. Under physiological conditions, there is no exogenous DNA in the cell, but when viral infections or cellular damage occur, exogenous DNA appears. At this point, cGAS synthesizes the cGAMP (cyclic GMP-AMP) dimer molecule. STING, located on the membrane of the endoplasmic reticulum (ER), serves as a receptor for cGAMP. Upon binding with cGAMP, STING undergoes a conformational change and translocates to the Golgi apparatus, where it activates downstream signaling molecules such as TBK1 and IRF3. This activation ultimately triggers the expression of type I interferons, pro-inflammatory cytokines, and other immune-related genes, helping the body resist pathogen invasion, eliminate damaged cells, and exert antiviral, antitumor, and immune regulatory effects [8].
The cGAS-STING pathway is not only a critical component of antiviral immunity but also plays a significant role in regulating various physiological and pathological processes, including cell death, tumor immunity, and anti-inflammatory responses. Therefore, understanding the molecular mechanisms underlying this pathway and elucidating its functions in different cells and tissues is crucial for the development of novel immunotherapies. Despite the increasing recognition of the role of the cGAS-STING pathway in disease defense, current research still faces several bottlenecks. These bottlenecks primarily include an incomplete understanding of the fine-tuned regulatory mechanisms that activate the pathway and challenges in developing targeted therapeutics [9]. Issues such as multiple variations of the STING receptor, the stability of cGAMP, and the complexity of the interaction between cGAS and STING limit the clinical application potential of this pathway [10].
It is important to note that the role of the cGAS-STING pathway extends beyond immune defense. It is also closely associated with the development and progression of various diseases, including cancer and autoimmune diseases. Therefore, research into the regulatory mechanisms of this pathway holds significant theoretical and clinical value in immunology, oncology, and other related fields.
This review aims to summarize the mechanisms of the cGAS-STING pathway in different physiological and pathological states and to analyze the challenges and bottlenecks it faces in clinical applications. By providing a comprehensive biological analysis of the cGAS-STING pathway, this paper seeks to offer new insights and targets for drug development in related diseases and to provide a theoretical foundation for the optimization of future therapeutic strategies. It is hoped that this review will inspire new breakthroughs and directions in the field of disease treatment, particularly in immunotherapy for cancer, autoimmune diseases, and viral infections.
Activation and Inhibition of the cGAS-STING Pathway
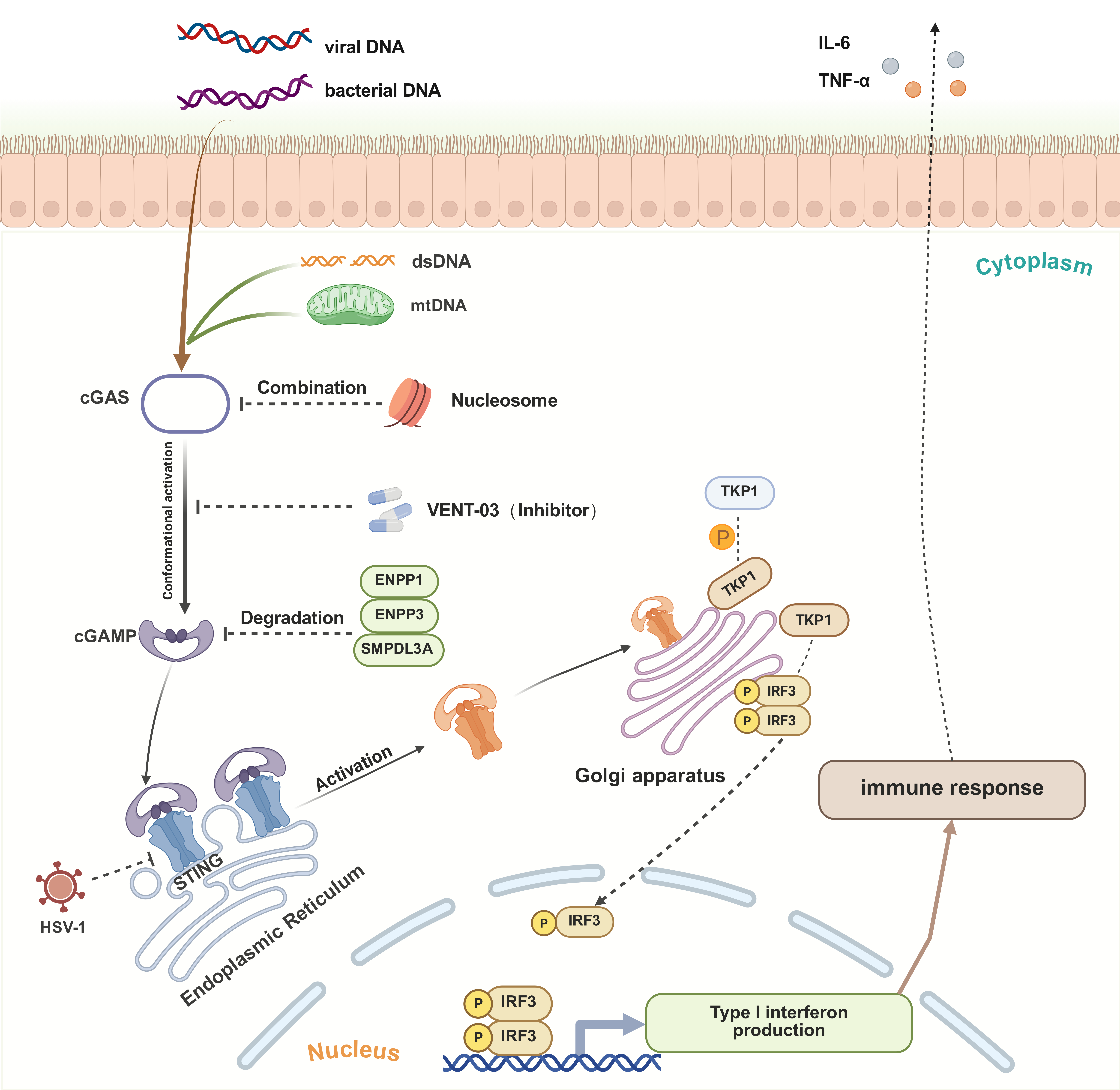
When the chromosomal DNA of a virus or cancer cell enters the cytoplasm and binds to cGAS, cGAS catalyzes the production of 2'3'-cGAMP. On the ER, STING undergoes a conformational change upon sensing 2'3'-cGAMP, causing the cGAS-STING signaling pathway to transition from an inactive (closed) state to an active (open) state. This transition promotes the assembly of the TBK1-IRF3 complex, ultimately triggering type I interferon responses and the release of other inflammatory cytokines [11, 12]. As a sentinel for pathogen invasion, cGAMP ensures the pathway remains activated until the virus or pathogen is cleared. Once eliminated, the cGAS-STING pathway reverts to its closed state. Studies have shown that cGAMP could be degraded by several enzymes, such as ENPP1, ENPP3, and SMPDL3A, to limit cGAS-STING signaling and maintain systemic inflammatory homeostasis [13].
Under physiological conditions, the activation and inhibition of the cGAS-STING pathway are tightly regulated. However, some viruses possess mechanisms to suppress this pathway. For instance, the HSV-1 virus inactivates STING, thereby inhibiting the cGAS-STING pathway to evade the innate antiviral immune response [14]. Additionally, methyltransferase PRMT6 has been shown to impair the TBK1-IRF3 signaling cascade, weakening the innate antiviral immune response [15]. These findings emphasize the crucial role of the cGAS-STING pathway in defending against viral invasion and activating the innate immune system.
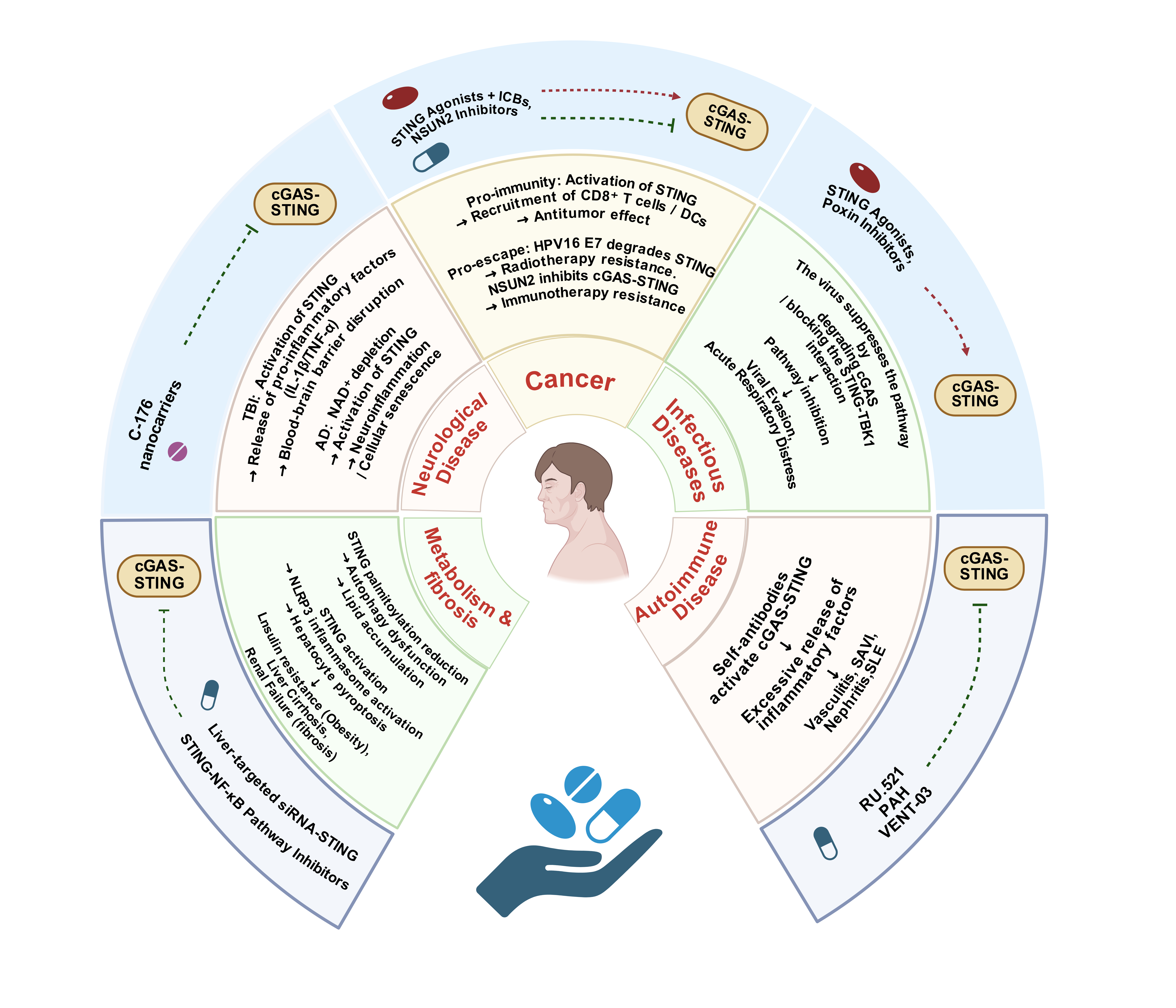
In certain pathological conditions, such as systemic lupus erythematosus (SLE), Stimulator of IFN Genes-Associated Vasculopathy with Onset in Infancy (SAVI), and systemic sclerosis, the accumulation of abnormal DNA could activate the cGAS-STING pathway, leading to sustained activation of downstream immune signaling and exacerbating inflammation-mediated damage [16]. The use of cGAS-specific small molecule inhibitors effectively suppresses interferon expression triggered by dsDNA, mitigating inflammation [17]. Examples of such inhibitors include RU.521 [18], PAH [19], and VENT-03 [20]. Notably, the VENT-03 inhibitor has entered Phase I clinical trials, representing a novel therapeutic approach for autoimmune diseases (Figure 1).
Figure 1. Fundamental Activation and Inhibition Mechanisms of the cGAS-STING Pathway.
Crosstalk Between the cGAS-STING Pathway and Subcellular Organelles
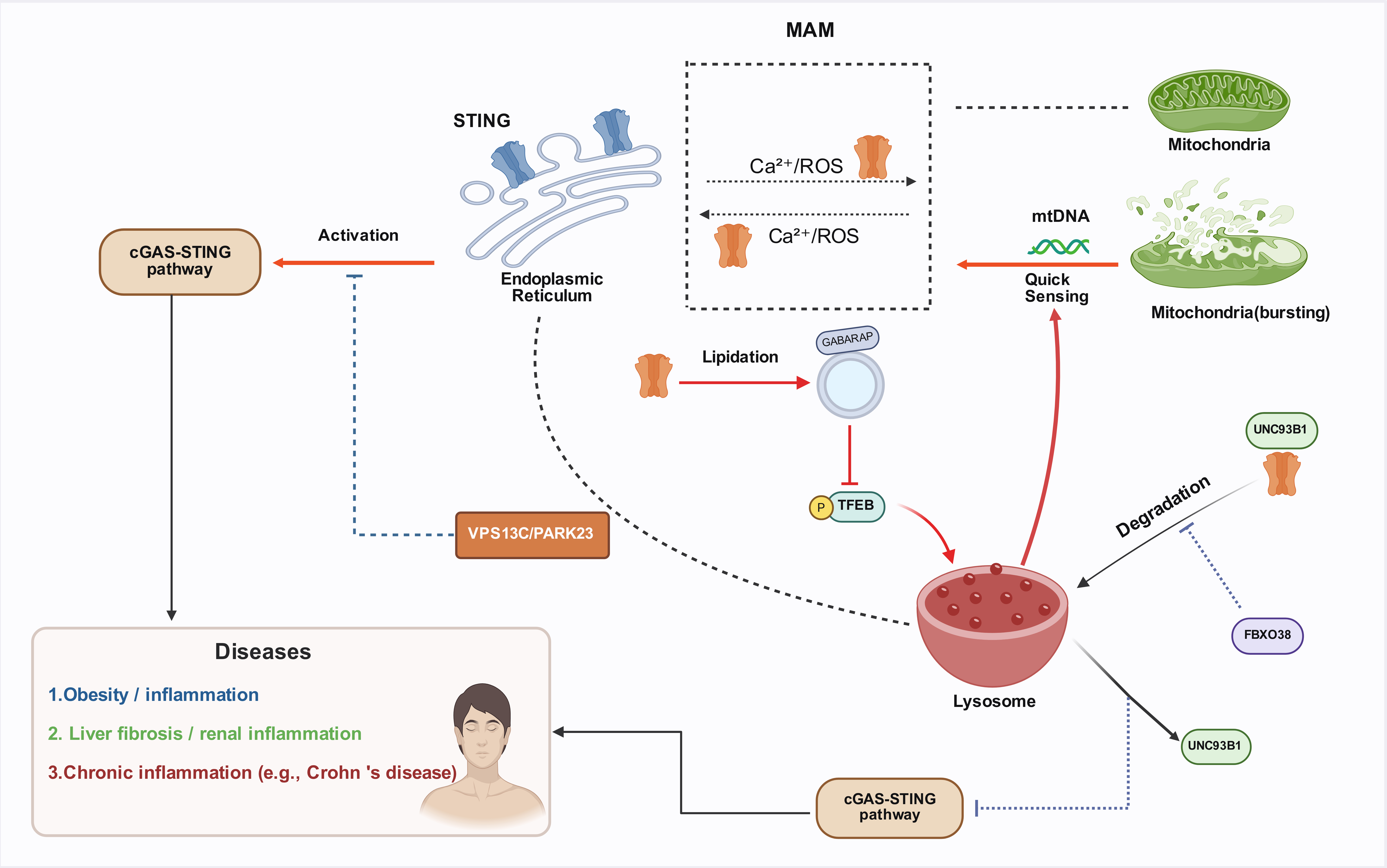
Regulation of STING by the ERSTING is primarily localized within the ER, where it can activate the NF-κB and IRF3 transcriptional pathways, thereby inducing the expression of type I interferons (e.g., IFN-α and IFN-β), which subsequently promote an effective antiviral state upon expression [21]. There is a close anatomical and functional connection between the ER and mitochondria, which communicate through calcium ions and reactive oxygen species (ROS), facilitating inter-organelle signaling. STING is highly localized in the ER-mitochondria-associated membrane (MAM) regions, a unique position that allows it to respond acutely to cellular organelle stress, such as the leakage of mitochondrial DNA (mtDNA) [22]. The ER plays a crucial role in protein folding, lipid synthesis, and calcium storage, serving as an important platform for STING synthesis, modification, and residence. The interaction between STING and the ER forms the core of the cGAS-STING pathway, making the ER a key hub in the regulation of STING signaling [23].
Crosstalk Between Lysosomes and STING SignalingStudies have shown that there is a reciprocal regulatory relationship between the cGAS-STING pathway and lysosomes. During pathogen invasion, the cGAS-STING pathway activates the transcription factor TFEB, which promotes lysosomal biogenesis and accelerates the clearance of cytosolic DNA and invading pathogens [24]. This process highlights that inducing lysosomal biogenesis is another important function of the cGAS-STING pathway. In a mouse model of HSV-1 infection, UNC93B1 targets STING to promote the autophagy-lysosome degradation pathway, which in turn reduces the activity of the cGAS-STING signaling pathway [25]. Furthermore, the absence of the T-cell immune-related FBXO38 protein leads to lysosome-dependent STING degradation, inhibiting the activation of the STING pathway [26].
Moreover, research indicates that the ER-lysosome lipid transporter VPS13C/PARK23 could inhibit abnormal mtDNA-dependent STING signaling [27]. Recent studies show that STING induces the lipidation of GABARAP on single-membrane vesicles, specifically inhibiting mTORC1-mediated phosphorylation of TFEB. Subsequently, TFEB translocates to the nucleus to regulate the expression of lysosome-related genes. STING-activated lysosomes not only efficiently clear cytosolic DNA but also enhance the clearance of bacteria (e.g., Salmonella Typhimurium) and viruses (e.g., HSV-1) [24]. These studies underscore the significant role of STING in inter-organelle interactions (Figure 2).
Figure 2. Crosstalk Mechanisms between the cGAS-STING Pathway and Subcellular Organelles (ER / Mitochondria / Lysosomes).
Non-Coding RNA Regulatory Networks
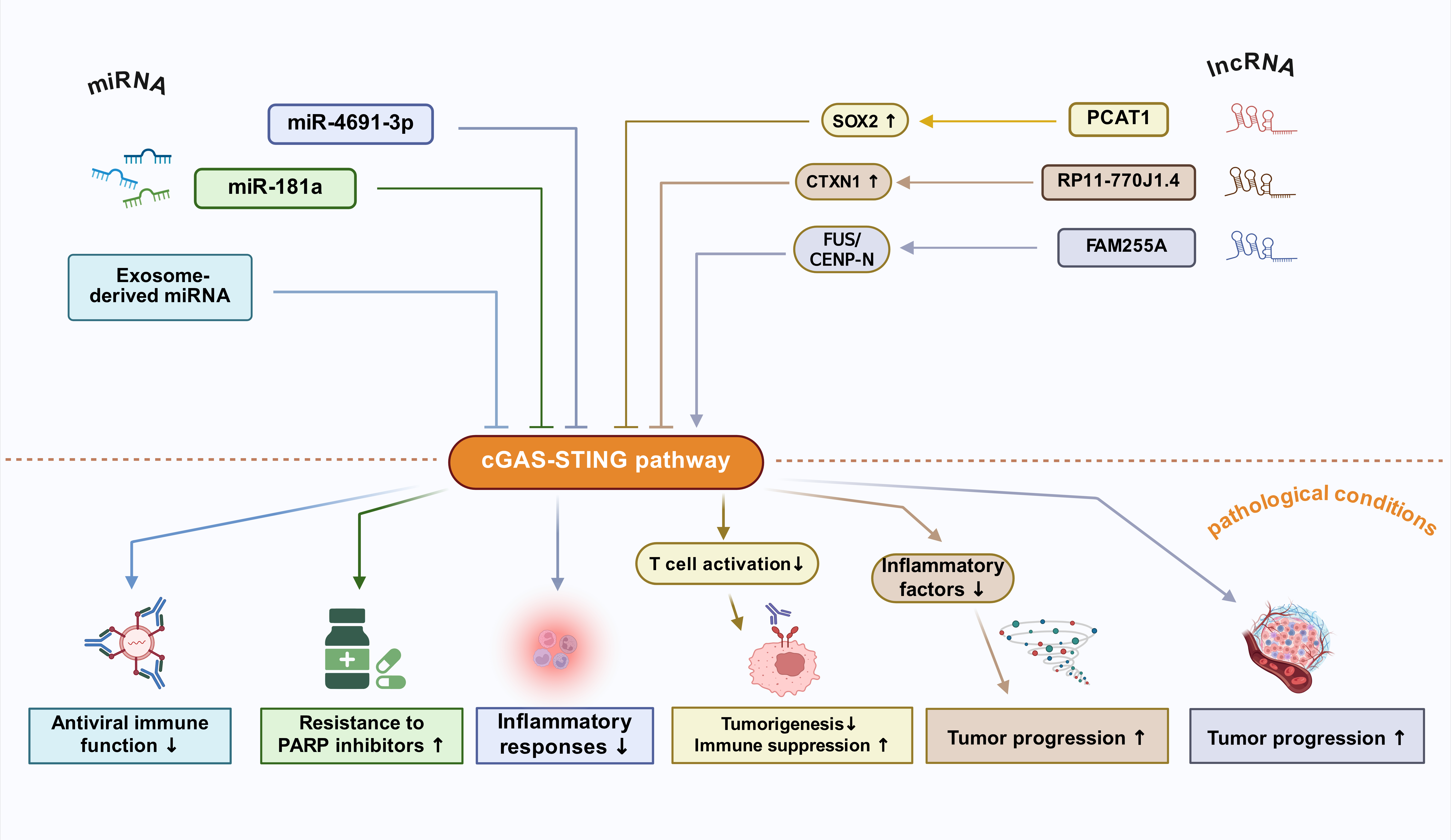
miRNA Regulation of STINGMicroRNAs (miRNAs) are non-coding RNA molecules, typically around 20-24 nucleotides in length, that play a crucial role in regulating STING gene expression [28]. Studies have shown that STING is a direct target of miR-4691-3p, which inhibits STING expression and negatively regulates the cGAS-STING pathway, thereby suppressing inflammatory responses [29]. Additionally, miR-181a can target STING to inhibit the production of pro-inflammatory factors, promoting resistance to PARP inhibitors in triple-negative breast cancer (TNBC) and ovarian cancer (OVCA) [28]. Similarly, in multiple myeloma (MM), exosome-derived miRNA secretion suppresses the antiviral immune function of the cGAS-STING pathway [30].
lncRNA Regulation of STINGStudies have shown that long non-coding RNAs (lncRNAs) are closely associated with the activation of the cGAS-STING pathway and play significant regulatory roles in both physiological and pathological processes [31]. In non-small cell lung cancer (NSCLC), lncRNA PCAT1 could inhibit T cell activation mediated by the cGAS-STING signaling pathway through the activation of SOX2, thereby promoting tumorigenesis and immune suppression [32]. In glioma, the inhibition of lncRNA RP11-770J1.4 downregulates the expression of the downstream protein CTXN1, activates the cGAS-STING pathway, and induces the secretion of related inflammatory factors [33]. In nasopharyngeal carcinoma (NPC), lncRNA FAM255A regulates the expression of CENP-N through interaction with FUS, affecting the cGAS-STING pathway. Specifically, the activation of the FUS/CENP-N/cGAS-STING signaling pathway promotes tumor progression, while the suppression of lncRNA FAM255A expression weakens the malignant characteristics of tumor cells [34].
These studies highlight the close regulatory association between non-coding RNAs and STING, suggesting that they may serve as upstream regulatory genes for STING and potential molecular targets for therapeutic interventions (Figure 3).
Figure 3. Regulation of the cGAS-STING pathway by non-coding RNAs (miRNA/lncRNA).
Inflammation-Related Diseases
cGAS-STING Pathway Regulation of Inflammation-Related MechanismsThe cGAS-STING pathway activates the expression of pro-inflammatory factors such as LPS, IL-6, and IL-1β through the non-classical NF-κB pathway, thereby promoting the exacerbation of the inflammatory response [35]. Studies have shown that the gut microbiota can initiate a systemic antiviral immune response through the cGAS-STING-IFN-I axis [36]. Additionally, autophagy regulates the cGAS-STING pathway negatively by clearing cytosolic DNA. Defects in this process may lead to the development of chronic inflammatory diseases such as Crohn’s disease [37]. At the molecular level, the TBK1-activated p62/SQSTM1- mediated autophagy pathway effectively weakens the transmission of cGAS-STING signals [38], thus modulating the intensity and duration of immune responses and preventing immune dysregulation and chronic inflammation caused by excessive activation. Meanwhile, STING activation also suppresses the secretion of the anti-inflammatory factor IL-10, further exacerbating the inflammatory response [39, 40]. However, there is controversy regarding STING's regulation of IL-10. Some studies suggest that STING activation can promote IL-10 secretion in certain inflammatory environments, especially in intestinal inflammation [41]. This suggests that the immunoregulatory role of STING may depend on specific physiological and pathological states.
Systemic Autoimmune DiseasesGain-of-function mutations in STING lead to excessive activation of the cGAS-STING pathway, triggering an overactive inflammatory response. For example, mutations in the TMEM173 gene (such as N154S and V155M) result in sustained STING activation, causing systemic autoimmune vasculitis and pulmonary fibrosis, known as SAVI, which typically manifests in infancy [42, 43]. Research has shown that interferon-stimulated genes (ISGs) are persistently upregulated in SAVI patients, closely associated with the overactivation of the JAK-STAT signaling pathway. This abnormal activation not only promotes enhanced immune responses but may also worsen the inflammatory process of autoimmune diseases, highlighting the central role of the cGAS-STING pathway in the pathogenesis of SAVI [44, 45].
Studies have indicated that genetic STING gain-of-function mutations are critical factors in familial inflammatory syndromes with lupus-like symptoms, revealing an important link between STING and SLE [46, 47]. It has been reported that self-antibodies in SLE patients continually activate the cGAS-STING pathway, resulting in the release of numerous inflammatory factors and further exacerbating the condition [48].
In lupus nephritis (LN), STING activation promotes ferroptosis and inflammation through the TBK1/NF-κB signaling pathway, advancing disease progression [49]. However, studies in mouse models have found that the cGAS-STING pathway does not promote autoimmune responses in SLE mouse models [50]. This finding suggests that directly applying mouse model findings to human diseases may present challenges due to the complex genetic background of SLE patients.
Organ-Specific InflammationIn the pathogenesis of alcoholic hepatitis, leakage of mitochondrial DNA (mtDNA) is considered a key trigger of immune responses. mtDNA activates the cGAS-STING pathway, thereby stimulating downstream IRF3 and NF-κB signaling pathways. The activation of these pathways leads to the excessive secretion of pro-inflammatory cytokines such as IL-6 and TNF-α, further aggravating the liver's inflammatory response. mtDNA leakage is closely related to alcohol-induced hepatocyte damage, and the cGAS-STING pathway plays a key role in the immune dysregulation and inflammatory response in alcoholic hepatitis [51, 52].
In LN, STING activation induces NLRP3 inflammasome activation and promotes necroptosis of kidney macrophages, thereby worsening the formation of proteinuria [53]. However, STING deficiency can alleviate symptoms of glomerulonephritis [54]. Mechanistically, STING upregulation enhances TBK1 expression and activates NF-κB signaling, which triggers ferroptosis and intensifies renal inflammation [49]. Therefore, regulating STING activity may represent a potential strategy for treating LN.
Studies have shown that under oxidative stress conditions, STING accelerates retinal pigment epithelial cell senescence through the NF-κB/HIF-1α signaling pathway [55]. In blood flow patterns, oscillatory shear stress (OSS) activates the ROS-STING axis, leading to endothelial cell senescence and promoting the development of atherosclerosis [56]. Furthermore, it has been shown that in aging endothelial cells, the cGAS-STING pathway is activated, further damaging vascular dilation function, while inhibition of cGAS-STING expression helps protect vascular function [57]. In Alzheimer's disease (AD), NAD+ depletion activates the cGAS-STING pathway, exacerbating neuroinflammation and accelerating cellular senescence. Supplementing NAD+ has been shown to alleviate cellular senescence effectively [58]. This suggests that the STING signaling pathway plays a critical role in the progression of age-related chronic diseases.
Future research directions include developing tissue-specific STING inhibitors, such as targeting kidney-specific nanoparticles to deliver H-151 inhibitors, to improve treatment targeting and efficacy [59]. Additionally, analyzing the structure-activity relationship of STING mutants (e.g., SAVI-related variants) will aid in the design of highly selective allosteric modulators to precisely regulate the cGAS-STING pathway, thereby improving the therapeutic outcomes for related diseases. These studies will provide new treatment strategies for clinical applications.
Infection and Immune Response
Antiviral ImmunityThe cGAS-STING pathway activates intracellular immune responses through the recognition of viral DNA (such as HSV-1), triggering the secretion of IFN-I [60]. This process depends on cGAS recognizing viral DNA to generate cGAMP, which activates the STING protein. The activated STING interacts with TBK1 to promote its phosphorylation, further activating the transcription factor IRF3, which ultimately induces the expression of IFN-I [61, 62].
RNA viruses, such as SARS-CoV-2 [63] and respiratory viruses (RVs) [64], induce mitochondrial dysfunction upon infection, leading to the release of mtDNA into the cytoplasm. Released mtDNA is recognized as a danger signal, further activating the cGAS-STING pathway. Following STING activation, a phosphorylation cascade involving TBK1 and IRF3 induces the production of a large amount of type I interferons and inflammatory factors, triggering a cytokine storm [65].
Studies have shown that HSV-1 escapes host immune surveillance by targeting the cGAS-STING pathway, inhibiting the immune response to the virus. STING is an important recognition molecule in the immune system that senses intracellular DNA damage or infection signals, activating downstream interferon responses and initiating antiviral immunity. HSV-1 effectively suppresses this immune response by disrupting the cGAS-STING pathway, facilitating its survival within the host [61]. Therefore, key regulation of the cGAS-STING pathway for oncolytic virus therapy may become an important strategy to improve efficacy. Optimizing cGAS-STING pathway activation or blocking its evasion mechanisms may enhance the immune effects of oncolytic viruses, highlighting the value of precise regulation of the cGAS-STING pathway in this therapeutic context [66].
However, some viruses can suppress host antiviral immune responses through various specific immune evasion strategies. For instance, the UL41 protein of herpesvirus degrades cGAS or blocks the binding of STING with TBK1, inhibiting IFN activation and thereby evading immune surveillance [67]. HPV11 targets STING for ubiquitin-mediated degradation via the E7 protein, reducing the expression of IFN-I in epithelial cells and further evading host immune defense [68]. However, adenoviruses have a minimal impact on this immune evasion mechanism, indicating that different viruses exhibit variability in their immune escape strategies [69].
In addition to collaborating with RIG-I to recognize RNA viruses, STING interacts with other PRRs to coordinate immune responses. For example, TBK1 recruits STING to activate IRF3 and NF-κB, mediating immune defense against tumors and viral infections [70]. Moreover, STING and RIG-I activate the IFN-I cascade via the mitochondrial adapter protein MAVS and TBK1, demonstrating the central role of STING in regulating host immune responses [71]. In summary, the cross-regulation between STING and PRR pathways amplifies the host's immune response to viruses and optimizes antiviral defense mechanisms.
Bacterial and Parasitic InfectionsThe cGAS-STING pathway recognizes the DNA or metabolic products of Legionella, inducing the production of IFN-I and pro-inflammatory cytokines to restrict bacterial replication. However, the HAQ-STING variant significantly weakens this immune response, increasing the host's susceptibility to Legionella [72]. Additionally, during the later stages of the developmental cycle, Chlamydia trachomatis activates STING through the CTL0390 protein, the key molecule that connects C. trachomatis to STING and mediates the 'STING-dependent lysis process.' This activation regulates the translocation of STING to the Golgi apparatus. Subsequent to the activation of STING, which leads to lytic expulsion, ultimately aiding the release of the pathogen from the host cell [73].
In sepsis-induced acute lung injury (ALI), the cGAS-STING pathway significantly enhances inflammation by activating the PARP-1/NLRP3 signaling pathway, leading to pathological damage in lung tissue, pulmonary edema, and exacerbated inflammation [74]. Furthermore, research indicates that STING deficiency aggravates Gram-negative bacterial infections, suggesting a complex bidirectional regulatory role for STING in immune responses [72]. Thus, the expression levels of STING may play a key "balancing" role in different pathological states, potentially promoting disease progression or, in some cases, inhibiting pathological processes.
Plasmodium infections activate the host immune response via the cGAS-STING pathway, inducing the production of IFN-I [75]. This process enhances the expansion of Treg cells, exerting immune-suppressive effects and limiting excessive inflammation and pathological damage. The activation of the cGAS-STING pathway not only enhances IFN-I expression but also promotes the proliferation of Treg cells, playing an important role in the immune regulation of Plasmodium infections [76].
Immune Evasion MechanismsStudies indicate that the NS4B protein of HCV can directly bind to STING and inhibit RIG-I-mediated IFN-I expression, helping the virus evade host immune responses [77]. Additionally, flaviviruses could activate the RIG-I-STING pathway, causing neuronal death and triggering inflammatory responses. This mechanism underscores the key role of this pathway in viral infections of the nervous system [78]. Moreover, poxviruses suppress IFN-I responses induced by dsDNA via the cGAS-STING pathway, inhibiting host immune responses by blocking STING activation, thereby regulating the cGAS-STING pathway to promote viral survival [79, 80].
STING's function is significantly influenced by genetic polymorphisms, such as R232, H232, and HAQ variants. For example, the H232 variant exhibits impaired function, resulting in increased susceptibility to DNA viruses like HSV-1 and MVA, while the HAQ and R232 variants maintain normal function [81]. Additionally, STING deficiency weakens monocyte differentiation and antigen-presenting capacity, affecting immune responses. In HIV-infected individuals, the HAQ/HAQ STING variant is associated with lower chronic immune activation and slower disease progression [82], suggesting that STING genetic variants may modulate immune responses and influence host susceptibility to viral infections.
Moreover, chronic viral infections, such as HIV, are often accompanied by prolonged immune activation. Although antiretroviral therapy (ART) effectively suppresses the virus, it may lead to long-term STING activation, thereby triggering autoimmune responses. In this context, the expression of STING and cGAS genes is significantly downregulated, while autoantibody production increases, indicating the important role of STING in maintaining immune tolerance [83]. Furthermore, activating mutations in STING1 can lead to SAVI, which presents with early-onset systemic inflammation, skin vasculopathy, and interstitial lung disease (ILD). Although the clinical features of SAVI are relatively well defined, its specific molecular mechanisms remain unclear and require further investigation to reveal STING's potential role in autoimmunity and related diseases [42].
Future research could focus on two main areas: first, the screening of broad-spectrum antiviral compounds, particularly those targeting the pathogen-STING interaction interface, such as poxin inhibitors from poxviruses, which could provide new strategies for antiviral therapy [84]. Secondly, a deeper understanding of the impact of STING allele polymorphisms (e.g., R232, H232, and HAQ) on susceptibility to infections across global populations will help elucidate the relationship between individual immune response differences and disease susceptibility, offering more efficient approaches for personalized immunotherapy [85].
Metabolism and Fibrosis
Metabolic AbnormalitiesObesity is widely regarded as a risk factor for various cancers and is closely associated with chronic inflammation. In adipocytes, mitochondrial dysfunction leads to mtDNA leakage, activating the cGAS-STING pathway, which reduces fat accumulation by promoting autophagy in adipocytes [86, 87]. Studies have shown that palmitoylation of STING plays a key role in the development of obesity. Fatty acid oxidation significantly inhibits the antiviral activity of STING by reducing its palmitoylation, a critical modification for activating its downstream signaling pathways. This inhibitory effect on fatty acid oxidation may impair STING's normal function by lowering palmitoylation [88]. Palmitoylation typically occurs in the Golgi apparatus, and its levels are significantly reduced in obesity models, resulting in abnormal binding between STING and TBK1, which suppresses its normal autophagic function, thereby exacerbating fat accumulation and promoting the development of obesity [89]. Additionally, the cGAS-STING pathway plays an anti-inflammatory role in adipocytes by promoting mitophagy, thus inhibiting excessive activation of the inflammatory response. Inhibition of STING expression leads to a reduction in autophagosome numbers, disrupting the balance of fat metabolism [86]. This process may exacerbate inflammation in adipocytes and further increase the risk of cancer associated with obesity. Therefore, STING may play a crucial role in the link between obesity and cancer, regulating metabolic and inflammatory responses in adipocytes. This suggests that dysregulated palmitoylation of STING could be an important mechanism in obesity-related metabolic disorders, and it indicates a close connection between lipid metabolism and immune responses. This provides a potential therapeutic target for future treatments targeting the cGAS-STING pathway.
Glucose could regulate the cGAS-STING pathway. Studies have shown that high glucose concentrations can induce STING activation, promoting macrophage polarization to the M1 type, thereby inhibiting wound healing in diabetic patients [90]. In type 2 diabetes models, reducing STING expression can improve peripheral insulin resistance and correct glucose intolerance abnormalities [91]. During tumor development, NSUN2 acts as a glucose sensor and inhibits the cGAS-STING signaling pathway, thereby promoting tumor progression and increasing immune therapy resistance. In contrast, inhibiting NSUN2 activity activates the cGAS-STING pathway, not only curbing tumor growth but also enhancing the effectiveness of immune therapy [92].
Organ FibrosisIn liver fibrosis research, the cGAS-STING pathway, as a DNA sensor located in the cytoplasm, has attracted significant attention. Studies have found that STING is expressed in non-parenchymal liver cells, particularly in macrophages [93]. In patients with non-alcoholic fatty liver disease (NAFLD), the expression of STING in monocyte-derived macrophages is closely related to the worsening of liver inflammation and fibrosis [94]. Furthermore, STING activates the NLRP3 inflammasome, inducing pyroptosis in hepatocytes and thereby exacerbating the progression of liver fibrosis [95]. These findings suggest that dysregulated STING expression may be a key driver of liver fibrosis progression.
Pulmonary fibrosis is a progressive and ultimately life-threatening lung disease. Studies have shown that abnormal activation of cGAS-STING participates in and promotes the development of fibrotic lung diseases. Polystyrene microplastics (PS-MPs) can promote ferroptosis in alveolar epithelial cells through cGAS-STING pathway, thereby triggering pulmonary fibrosis [96, 97]. However, other studies have pointed out that in idiopathic pulmonary fibrosis (IPF), STING has a protective effect on lung fibrosis, with its reduced expression exacerbating the fibrotic process [98].
Renal fibrosis is a common lesion leading to end-stage renal failure. Studies have shown that activation of the STING/ACSL4 pathway promotes ferroptosis and inflammation, further advancing chronic kidney disease (CKD) [99]. Additionally, mitochondrial damage and activation of the cGAS-STING pathway exacerbate kidney inflammation and fibrosis progression [100]. Butyrate, through modulation of the STING/NF-κB/p3 pathway, can affect NLRP65-mediated pyroptosis, thereby alleviating kidney fibrosis symptoms in CKD patients [101].
Future research could focus on developing tissue-specific regulation strategies for the STING pathway, such as using liver-targeted lipid nanoparticles (LNPs) to deliver siRNA-STING, to explore their effects on liver metabolism and fibrosis [102]. Additionally, establishing multi-omics integration platforms to analyze the dynamic network of the metabolism-fibrosis-immune axis could provide a more comprehensive understanding of the mechanisms, help identify new therapeutic targets, and promote the application of precision medicine in metabolic diseases [65, 103]. These studies are expected to reveal the role of the cGAS-STING pathway in various diseases and provide new ideas for clinical interventions.
cGAS-STING Pathway and the Nervous System
cGAS-STING Pathway Regulates NeuroinflammationStudies have shown that under hypoxic conditions, glioblastoma (GBM) cells release extracellular vesicles (EVs) carrying miR-25/93 to macrophages, thereby inhibiting the cGAS-STING pathway, reducing type I interferon secretion (e.g., IFN-β), lowering the expression of M1 polarization-related genes (e.g., Cxcl9, Cxcl10, Il12b), and weakening macrophage anti-tumor immunity and T cell activation, which further fosters an immunosuppressive tumor microenvironment (TME) [104]. Traumatic brain injury (TBI) triggers the activation of the cGAS-STING pathway, which exacerbates neuroinflammatory responses through type I interferons (IFN-α/β) and pro-inflammatory factors (e.g., TNF-α, IL-1β, and IL-6), while also inducing autophagy dysfunction (e.g., abnormal LAMP2). Studies have shown that STING gene knockout (STING⁻/⁻) could reduce the release of inflammatory factors, decrease lesion volume, and restore autophagic function, suggesting that STING exacerbates neuroinflammatory damage by enhancing type I interferon signaling [105].
In a mouse spinal cord injury model, STING interacts with TBK1 to enhance TBK1 phosphorylation, activating downstream NF-κB and MAPK signaling pathways that amplify the inflammatory response of microglial cells, whereas suppressing STING expression reduces the activation of these pathways and alleviates the inflammatory response, thereby facilitating spinal cord injury repair [106, 107]. This suggests that STING may play a role in spinal cord injury by regulating inflammatory responses.
In HSV-1 encephalitis, neurons promote the secretion of IFN-λ via the activation of the cGAS-STING pathway, which aids in antiviral immunity and suppresses viral spread [61, 108]. However, a study indicates that excessive activation of STING may trigger an overactive inflammatory response, leading to blood-brain barrier disruption, thus exacerbating neuronal damage and disease progression [109]. Therefore, the regulation of the cGAS-STING pathway needs to be finely balanced to ensure defense against viral infections while preventing damage to the blood-brain barrier.
Non-Classical Regulation of Neuronal Function by STINGIn addition to its regulatory functions through the classical cGAS-STING pathway, STING could regulate neuronal functions through non-classical pathways. Research suggests that intestinal neuroglial cells may employ alternative signaling mechanisms or express STING solely under certain disease conditions, with studies also revealing potential pathways for neuroglial cell-microbe communication within the intestinal nervous system [110, 111]. In a multiple sclerosis (MS) model, STING is activated in neurons and triggers the non-classical STIM1-STING signaling pathway, leading to the autophagic degradation of glutathione peroxidase 4 (GPX4) and causing ferroptosis [112]. This initiates inflammatory stress responses and cell death in neurons. STING is indirectly regulated by the biological clock gene BMAL1 through the LINE1-cGAS-STING pathway. When BMAL1 is deficient, heterochromatin stability is reduced, LINE1 is aberrantly activated, and the cGAS-STING pathway is triggered, leading to type I interferon responses and the senescence-associated secretory phenotype (SASP) [113, 114]. This suggests that BMAL1, through its non-classical chromatin regulatory function, suppresses the LINE1-STING axis, maintains cellular homeostasis and delays aging.
STING Interaction with GBMResearch indicates that in GBM models, the activation of STING induces a strong immune response, mediates the NK cell-mediated tumor regression, and contributes to TME remodeling [115]. Additionally, preclinical studies have found that activation of the cGAS-STING pathway in the neuro-GBM immune microenvironment plays a positive role in therapy and achieves anti-tumor effects [116]. However, other studies have shown that in high-risk, recurrent-grade gliomas, STING expression is significantly upregulated, which may reflect the tumor cells' resistance to its effects [117].
Future research may explore the development of STING inhibitors that can cross the blood-brain barrier, such as utilizing nano-carriers for delivering C-176 analogs, to precisely regulate cGAS-STING pathway activity and mitigate or slow the progression of neurodegenerative diseases [66]. Additionally, exploring the role of STING in regulating various neurological diseases could open avenues for its application in personalized treatment. Another key direction is the study of combination therapies, such as combining STING modulation with immune agents, to explore synergistic effects in tumor suppression [118]. Furthermore, STING, as a potential biomarker for neurodegenerative diseases, warrants further exploration for early diagnosis and monitoring of these diseases [119].
Cancer and Immunotherapy
cGAS-STING Pathway and Tumor ImmunityAs the global incidence of cancer increases, tumors have become one of the leading causes of death, so timely diagnosis and intervention are crucial for improving cure rates and enhancing patients' quality of life [120, 121]. The cGAS-STING pathway serves as a crucial immune surveillance mechanism, inducing the production of IFN-I and various chemokines. These factors play a vital role in the recruitment and activation of CD8+ T cells [122]. In the TME, STING activation enhances immune responses, particularly the infiltration of T cells, contributing to its antitumor effects [123, 124]. Activation of the cGAS-STING pathway effectively suppresses the expression of immune checkpoint molecules such as PD-L1, thereby alleviating the immune suppression of tumor cells on T cells and promoting tumor immune clearance [125].
In certain subtypes of gastric cancer, such as dMMR/MSI-H gastric cancer, high STING expression has been confirmed to be closely associated with T cell infiltration. Studies show that patients with dMMR/MSI-H gastric cancer exhibit stronger immune responses and higher levels of T cell infiltration upon activation of the STING pathway, suggesting that these patients may benefit more from immune checkpoint inhibitor therapies [126, 127]. Based on this, STING emerges as a potential therapeutic target, capable of significantly enhancing the efficacy of immunotherapy and improving patient survival by enhancing immune responses and remodeling the TME [128].
cGAS-STING Pathway and Synergy with Immune CellsThe synergy between STING and innate immune cells plays a crucial role in tumor immunity. Dendritic cells (DCs), upon activation of STING, promote cross-presentation of tumor antigens through the cGAS-STING pathway, thereby enhancing antitumor immune responses [129]. This process not only enhances the immunogenicity of DCs but also strengthens the activation and functionality of T cells, facilitating the effective recognition and elimination of tumor cells. Research indicates that STING activation plays a key role in DC maturation, cytokine release, and the inhibition of immune escape, providing important support for the development of cancer immunotherapy [130, 131].
However, some studies have shown that in pancreatic cancer, STING agonists could inhibit NK cell antitumor activity by activating Breg cells to release IL-35, revealing the limitations of STING agonist monotherapy [132].
STING Suppression and Resistance to Targeted TherapySTING suppression is a significant mechanism of resistance to targeted cancer therapies. HPV16 E7 inhibits STING by promoting its degradation, thereby blocking the IFN-I signaling pathway and suppressing the antitumor immune response in cervical cancer cells. This enhances tumor resistance to radiotherapy, allowing tumor cells to evade host immune surveillance and increasing resistance to radiation therapy [133, 134]. Furthermore, in triple-negative breast cancer (TNBC), ARAh silences STING through epigenetic mechanisms, thereby inhibiting its immune response activation and diminishing the effectiveness of PARP inhibitors [135, 136]. These findings underscore the pivotal role of STING suppression in tumor immune evasion and therapy resistance, emphasizing the need for targeted therapies to modulate the STING pathway.
cGAS-STING Pathway and Oncogenic SignalingThe cGAS-STING pathway plays a double-edged sword role in cancer development. mtDNA leakage caused by mitochondrial damage activates the cGAS-STING pathway, triggering intracellular inflammation and promoting chromosomal instability (CIN) [137, 138]. This process supports tumor cell survival and accelerates tumor progression via an IL-6-dependent pathway. In BRCA1-deficient ovarian cancer, the STING-mediated inflammatory microenvironment further promotes immune evasion and weakens the antitumor immune response. Studies have shown that PARP inhibitors significantly reverse this immune escape phenomenon, aiding the immune system in recognizing and clearing tumor cells, thereby improving therapeutic outcomes [139]. Activation of the STING-TBK1 axis promotes the expression of ATP-citrate lyase (ACLY), enhancing fatty acid synthesis and driving macrophages toward M2 polarization by remodeling lipid metabolism. This chromatin-regulated process further affects immune cell metabolism and function [140, 141].
Dual Roles of STING in CancersSTAT3 deficiency disrupts the cGAS-STING-IFN pathway, thereby impairing the inhibitory effect of NK and NKT cells on SCLC metastasis and dissemination. This process can be restored through the overexpression of IRF7 or exogenous supplementation of IFN, thereby improving the prognosis of SCLC patients [142]. However, a study suggests that STING, through the TBK1-NF-κB pathway, contributes to the formation of an inflammatory microenvironment that promotes bone metastasis in cervical cancer [143]. Moreover, in recurrent gliomas, overexpression of STING correlates with IDH1 mutations, suggesting that STING may serve as an independent prognostic marker for glioma progression [144, 145]. These studies reveal the complex role of STING signaling in different cancer types.
Future research should focus on understanding the spatiotemporal activation mechanisms of STING in both tumor and immune cells. Single-cell spatial transcriptomics could particularly reveal its role within the TME [146]. Additionally, exploring STING agonists in combination with immune checkpoint inhibitors (ICIs) or IL-35 in combination therapies will help improve the effectiveness of antitumor immune responses, providing more effective treatment options in clinical therapy [132, 147].
Concluding Remarks
The cGAS-STING pathway is a crucial intracellular immune signaling pathway, primarily involved in the host's immune response to exogenous pathogens (such as viruses and bacteria) and responses to endogenous damage. Recent studies have shown that, in response to viral and pathogen invasion, STING plays a pivotal regulatory role in immune responses through various post-translational modifications (PTMs). The development of specific probes targeting different PTM states of STING could precisely regulate its activity, thereby enhancing the activation of the downstream TBK1-IRF3 pathway. For example, TMED2, in combination with the MITA signaling mediator, can further enhance IRF3 activation, improving the efficiency of antiviral immune responses [148].
As a key immune response regulatory mechanism, the cGAS-STING pathway is involved in the regulation of several subcellular organelles, such as the ER and lysosomes, and interacts with other intracellular signaling pathways to form a complex regulatory network. These organelles play an increasingly important role in cellular immune responses, inflammatory reactions, and pathogen defense, and their dysfunction is often closely associated with the development of various diseases. Therefore, exploring how to regulate the functions of these subcellular organelles via the cGAS-STING pathway offers new perspectives and possibilities for developing nanotechnology-based disease therapies. Nanotechnology can precisely target these intracellular structures, and by activating or inhibiting the cGAS-STING pathway, it can influence immune responses, opening up new frontiers in disease treatment.
On the other hand, non-coding RNAs (ncRNAs), which have been a focal point of research in recent years due to their significant roles in gene expression regulation, genome stability maintenance, and cellular physiological functions, have attracted widespread attention. NcRNAs play crucial roles in biological development and health maintenance and are also closely linked to the onset of various diseases. Increasing evidence indicates that ncRNAs regulate the activation and inhibition of the cGAS-STING pathway through direct or indirect interactions at various levels. For instance, certain microRNAs and long non-coding RNAs can modulate the expression or stability of cGAS or STING through interactions, thereby affecting the strength and duration of downstream immune responses. These findings provide a theoretical basis for regulating the cGAS-STING pathway via ncRNAs and offer new insights for the development of novel disease treatment strategies, such as gene therapy and immunotherapy. With the integration of nanotechnology, future approaches may precisely regulate the cGAS-STING pathway through targeting ncRNAs, achieving more refined therapeutic outcomes for diseases.
In recent years, the role of the cGAS-STING pathway in autoimmune diseases has increasingly attracted the attention of researchers. However, excessive or abnormal activation of the cGAS-STING pathway has been found to be closely associated with the onset of various autoimmune diseases, such as SLE, Sjögren’s syndrome, and rheumatoid arthritis. Studies have shown that when the activation of the cGAS-STING pathway becomes uncontrolled, it may lead to the loss of immune tolerance, triggering autoimmune responses that result in tissue damage and inflammation. Notably, in certain autoimmune disease patients, abnormal activation of the cGAS-STING pathway can promote the excessive secretion of inflammatory cytokines, thereby exacerbating the clinical symptoms of the disease.
To address this issue, researchers have been developing inhibitors of the cGAS-STING pathway as potential therapeutic strategies. Inhibitors such as VENT-03 and PAH have been found to effectively suppress the abnormal activation of the cGAS-STING pathway. Notably, the oral drug VENT-03 has completed its Phase I clinical trial and is scheduled to initiate Phase II clinical trials soon; it is expected to be used in the treatment of patients with SLE in the future. By inhibiting the cGAS-STING pathway, these inhibitors can significantly alleviate inflammation caused by excessive immune responses and reduce tissue damage, thereby mitigating the symptoms of autoimmune diseases. Particularly in the treatment of diseases such as SLE, inhibition of the cGAS-STING pathway is considered a promising strategy, as the abnormal activation of this pathway plays a key role in the pathogenesis of these diseases.
In cancer treatment, activating the cGAS-STING pathway to counter tumor progression has become a theoretically feasible approach. However, studies have found that the efficacy of using cGAS-STING agonists to combat tumor progression is suboptimal [149], and some research even suggests that it may further promote tumor progression, highlighting the limitations of cGAS-STING agonist monotherapy [132, 150]. Activation of the cGAS-STING pathway is crucial for initiating the initial anti-tumor immune response. Recent studies have shown that persistent activation of the cGAS-STING pathway in tumors could induce an immune-suppressive TME, promoting the survival and metastasis of cancer cells. Additionally, activation of the cGAS-STING pathway in myeloid-derived suppressor cells (MDSCs) has been shown to participate in the recruitment of MDSCs and enhance their immunosuppressive activity, thereby promoting TME remodeling [151–153]. Furthermore, while the cGAS-STING pathway may play an anti-tumor role in the early stages of cancer, tumor cells exhibit strong immune evasion capabilities. In the later stages of tumor development, the pathway could evolve into a chronic inflammatory state. Persistent inflammation could induce immune tolerance through mechanisms such as immune cell exhaustion, the expansion of regulatory cells, and clonal anergy, thus driving tumor progression [154, 155]. This suggests that using cGAS-STING agonists alone to combat tumor progression may not be the optimal approach. Furthermore, the mechanisms by which sustained activation of the cGAS-STING pathway promotes tumor progression remain a major research question. Additionally, current research on the cGAS-STING pathway in non-tumor cells of the TME is limited, and the role of this pathway in non-tumor cells remains unclear. These gaps hinder our deeper understanding of tumor mechanisms.
The rise of immunotherapy has opened a new chapter in cancer treatment, and the use of cGAS-STING agonists in combination with ICIs may represent a novel therapeutic approach in cancer therapy. A thorough investigation of the mechanisms underlying the cGAS-STING pathway in cancer therapy is crucial for advancing personalized treatment approaches, which holds significant clinical implications for deepening our exploration of cancer treatments [156, 157].
Moreover, in certain diseases, aberrant activation or inhibition of the cGAS-STING pathway is not only closely associated with the onset of the disease but also plays a crucial role in disease progression. For instance, in some autoimmune and chronic inflammatory diseases, excessive activation of the cGAS-STING pathway may lead to an overactive immune response, resulting in tissue damage and pathological changes. In certain viral infections, defects or inhibition of the cGAS-STING pathway could impair the host’s immune defenses, enabling persistent viral presence. Therefore, the precise modulation of the cGAS-STING pathway using specific agonists or inhibitors has become a critical strategy in treating these diseases. By regulating the activity of the cGAS-STING, it is possible to maintain immune defense while avoiding the side effects of excessive immune responses, thereby effectively treating autoimmune or chronic inflammatory diseases.
In conclusion, as a central pathway in the innate immune system, the cGAS-STING pathway plays an irreplaceable role not only in combating exogenous pathogens but also in cancer immunity, infections, inflammation, and autoimmune diseases. In-depth studies on the role of the cGAS-STING pathway in various diseases will lead to more precise targeted therapies for clinical treatment, promote the development of personalized medicine, and provide patients with additional treatment options. Therefore, investigating how to regulate the cGAS-STING pathway across diverse disease contexts will be a pivotal direction in future therapeutic research.
Abbreviations
Acute Lung Injury: ALI; Alzheimer's Disease: AD; ATP-Citrate Lyase: ACLY; Cyclic GMP-AMP: cGAMP; Cyclic GMP-AMP Synthase: cGAS; Dendritic Cells: DCs; Double-Stranded DNA: dsDNA; Endoplasmic Reticulum: ER; Extracellular Vesicles: EVs; Glioblastoma: GBM; Hepatitis C Virus: HCV; Herpes Simplex Virus Type 1: HSV-1; Human Papillomavirus Type 11: HPV11; Hypoxia-Inducible Factor-1α: HIF-1α; Immune Checkpoint Inhibitors: ICBs; Interferon Regulatory Factor 3: IRF3; Interferon-Stimulated Genes: ISGs; Lupus Nephritis: LN; Long Non-Coding RNA: lncRNA; Lipid Nanoparticle: LNP; Mi-tochondrial DNA: mtDNA; Myeloid-Derived Suppressor Cells: MDSCs; Non-Alcoholic Fatty Liver Disease: NAFLD; Nuclear Factor Kappa-B: NF-κB; NOD-Like Receptor Pyrin Domain-Con-taining Protein 3: NLRP3; Non-Small Cell Lung Cancer: NSCLC; Ovarian Cancer: OVCA; Programmed Death-Ligand 1: PD-L1; Regulatory T Cell: Treg; Stimulator of Interferon Genes: STING; Stimulator of Interferon Genes-Associated Vasculopathy with Onset in Infancy: SAVI; Systemic Lupus Erythematosus: SLE; TANK-Binding Kinase 1: TBK1; Traumatic Brain Injury: TBI; Tu-mor Microenvironment: TME; Triple-Negative Breast Cancer: TNBC; Type I Interferon: IFN-I.
Declarations
Author Contributions
Yunyong Wang: Conceptualization, Writing -- original draft; Xiaohang Lu: Writing -- original draft; Jinna Tan: Writing -- re-view & editing; Jiaqian He: Writing -- review & editing; Hui Yin: Writing -- review & editing; Peipei Chen: Visualization; Hemeng Wu: Visualization; Yuzhen Luo: Visualization; Mingfen Li: Su-pervision. All authors read and approved the final manuscript.
Acknowledgments
Not Applicable.
Funding Information
This work was supported by the Natural Science Foundation of Guangxi (Grant No. 2025GXNSFDA069035, 2025GXNS-FAA069372).
Ethics Approval and Consent to Participate
Not Applicable.
Competing Interests
The authors declare that they have no existing or potential commercial or financial relationships that could create a con-flict of interest at the time of conducting this study.
Data Availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
 Figures
Figures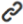 References
References Peer
Peer Information
InformationFigure 1. Fundamental Activation and Inhibition Mechanisms of the cGAS-STING Pathway.
Figure 2. Crosstalk Mechanisms between the cGAS-STING Pathway and Subcellular Organelles (ER / Mitochondria / Lysosomes).
Figure 3. Regulation of the cGAS-STING pathway by non-coding RNAs (miRNA/lncRNA).
Peer-review Terminology
Identity transparency: Single anonymized
Reviewer interacts with: Editor
Details
This is an open access article under the terms of the Creative Commons Attribution License(http://creativecommons.org/licenses/by/4.0/), which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Publication History
Received 2025-10-09
Accepted 2025-11-06
Published 2025-12-18