




Abstract
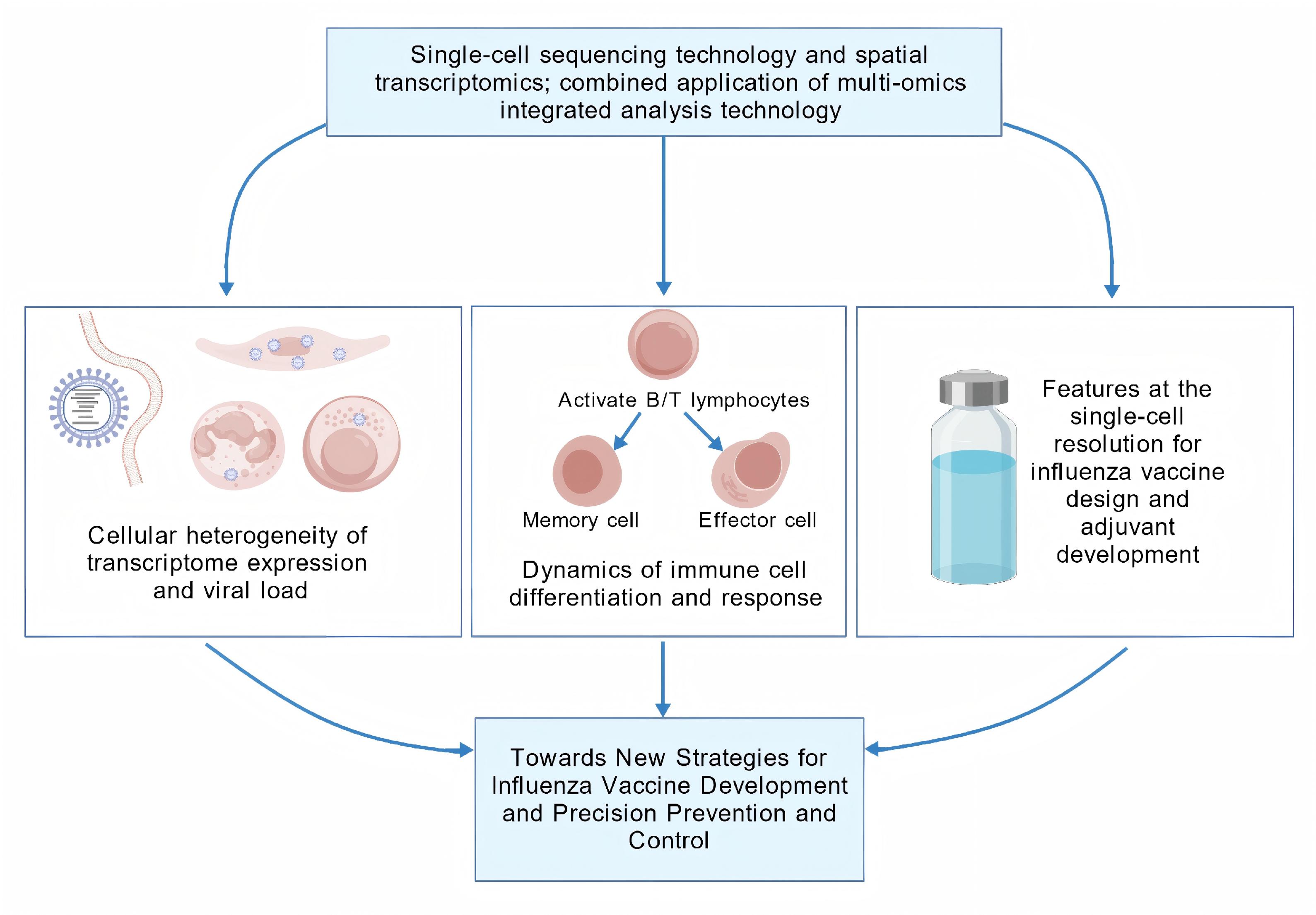
The integration of single-cell sequencing with spatial transcriptomics and multi-omics analyses has enabled a paradigm shift in biomedical re-search, thereby expanding its applicability and scientific impact. In the context of influenza virus studies, this technology has been instrumental in dissecting cellular heterogeneity, as demonstrated by its capacity to analyze differential transcriptomic profiles and reconstruct differentiation trajectories at the single-cell level following viral infection. These advances have provided mechanistic insights and a holistic understanding of influenza pathogenesis, surpassing the limitations of bulk-level analyses. This review provides a comprehensive dissection of cutting-edge appli-cations of single-cell sequencing in elucidating influenza virus infection mechanisms, immune cell heterogeneity, and vaccine development. By highlighting the single-cell resolution of virus–host interactions and vaccine efficacy studies, this work offers novel perspectives for designing precision-targeted antiviral interventions.
Keywords: Single-cell sequencing technology; Influenza virus; Cellular heterogeneity; Immune cell subset differentiation; Influenza vaccine.
Introduction
The genetic material of the influenza virus exhibits a high propensity for mutation, and genetic recombination can readily give rise to novel viral variants, leading to the emergence of new influenza strains [1]. This significantly hinders in-depth research on specific strains, thereby intensifying the challenges of vaccine development and drug screening [2]. Furthermore, influenza virus infection is prone to induce secondary infections and polymicrobial co-infections, the mechanisms of which are highly complex and multifaceted [3]. The severity and clinical outcomes of influenza virus infections are determined by a combination of viral and host factors. The multifactorial nature of the host immune response significantly influences the progression and severity of influenza virus infections, thereby substantially increasing the complexity of related research [4,5]. Influenza viruses employ a diverse array of strategies to evade host innate and adaptive immune responses. The multifaceted nature of their immune evasion mechanisms poses significant challenges in achieving a comprehensive understanding of the intricate virus–host immune system interactions [6,7]. Viral infections involve complex interactions with the structural and functional components of host cells, encompassing a multitude of molecular and cellular mechanisms [8]. In conclusion, addressing the aforementioned research challenges requires the implementation of high-precision experimental techniques and interdisciplinary collaborations.
Previous research methodologies have demonstrated limitations in elucidating the infection process and pathogenic mechanisms of influenza virus, particularly in accurately analyzing cellular heterogeneity and the complexity of virus–host interactions.For instance, conventional bulk sequencing techniques are incapable of resolving cellular heterogeneity in influenza virus infections; they also fail to detect rare cell subpopulations, gene expression variability, or mutational diversity, resulting in the loss of critical biological information [9]. Moreover, it is incapable of tracing the continuous trajectory of cellular state transitions, such as differentiation, development, or disease progression, rendering it unsuitable for rare cell populations (e.g., circulating tumor cells, early embryonic cells) or minimal clinical samples (e.g., needle biopsies) [10]. Moreover, mixed-cell sequencing fails to distinguish cell type- or subpopulation-specific gene expression patterns or mutational profiles, and is incapable of unbiased identification of novel cellular subpopulations [11]. In response to these challenges, the emergence of single-cell sequencing (SCS) technology has provided an effective solution by delivering high-resolution cellular-level data, thereby facilitating a comprehensive understanding of the impact of influenza virus infection on host cells [12]. The integration of multi-omics technologies has significantly advanced our understanding of gene regulatory networks during influenza virus infection, offering critical insights into how the virus evades immune surveillance and enhances viral replication by modulating the host cell transcriptomic profile [13,14]. Furthermore, compared to conventional bulk sequencing methodologies, SCS technology enables the identification of intercellular heterogeneity, which is crucial for elucidating the mechanisms of viral transmission and replication across diverse cell types [14]. This paper aims to systematically summarize the advancements in the application of SCS technology in influenza virus research, elucidate the significance of SCS technology in investigating infection mechanisms, reveal the single-cell resolution features of virus–host interactions and vaccine development, and offer novel perspectives on precision-based antiviral strategies.
Single-cell sequencing technology
The integration of SCS technology with multi-omics approaches facilitates a comprehensive analysis of the genome, transcriptome, and epigenome at the single-cell level, allowing for a more holistic understanding of cellular function and regulation [15]. Traditional sequencing techniques are performed at the multicellular level, producing averaged signal outputs across a cell population, and consequently masking variations in cellular heterogeneity (i.e., differences among individual cells). In influenza virus research, single-cell RNA sequencing (scRNA-seq) is widely employed as a key component of SCS technology, primarily used to investigate transcriptomic expression profiles at the individual cell level [16,17]. The experimental protocol consists of the following sequential steps: single-cell isolation, cell lysis and nucleic acid extraction, reverse transcription for cDNA synthesis, whole transcriptome amplification, library preparation, high-throughput sequencing, and bioinformatics analysis, including quality control, alignment, gene expression quantification, and cell clustering, to characterize cellular heterogeneity [18](Figure 1).
Figure 1. The experimental workflow of scRNA-seq technology involves single-cell isolation and capture, followed by nucleic acid amplification and library construction, selection of an appropriate high-throughput sequencing platform, and concludes with data processing and bioinformatics analysis.
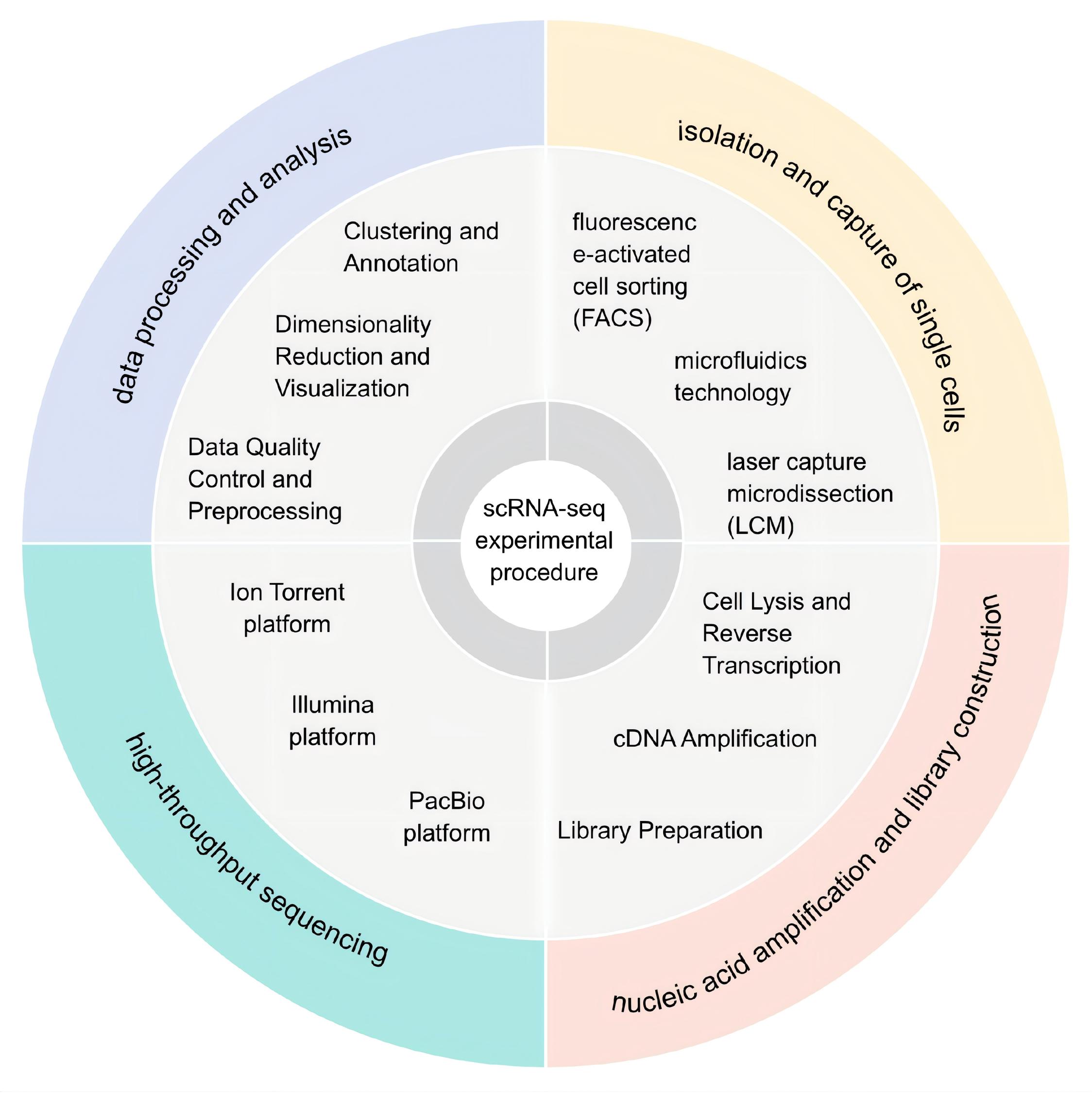
Depending on the research objectives, cells are isolated from tissues, cell lines, or body fluid. After dissociation into a single-cell suspension, automated microscopy or flow cytometry is used for cell quantification, along with a comprehensive evaluation of cell viability and quality parameters [19]. Subsequently, single-cell isolation is performed using conventional methods such as serial dilution, micromanipulation, and laser capture microdissection [20-22]. Currently, the most widely used techniques are Fluorescence-Activated Cell Sorting (FACS) and microfluidic-based platforms. FACS plays a crucial role in single-cell B-cell receptor/T-cell receptor sequencing (scBCR-seq/scTCR-seq) protocols, particularly when prior selection of specific cell types is necessary. This technology allows for the enrichment of defined B/T cell subpopulations and improves sample quality, serving as an essential tool in advanced immunological research [23,24]. Microfluidic technology enables precise manipulation of small liquid volumes via microchannels and microfluidic control components, leveraging microstructures such as micropores, microvalves, or droplet generators to compartmentalize cells into isolated reaction units (e.g., droplets or microchambers). This approach achieves specific functional outcomes through controlled droplet generation and the deliberate design of micropore architectures [25,26]. Droplet-based microfluidics technology, including methodologies such as Droplet-based single-cell RNA sequencing (Drop-seq) and 10x Genomics, integrates single-cell isolation and labeling into a streamlined workflow, enabling the parallel processing of thousands of cells. This advanced technique allows for precise control and facilitates the accurate separation and manipulation of individual cells [27-29]. Micro-well array technology (such as Sequencing Well) utilizes micro-well structures to enable the precise physical isolation of single cells, exhibiting comparable performance in accurate cell separation. In contrast, droplet-based microfluidics is better suited for applications that require ultra-high-throughput processing [30].
Nucleic Acid Amplification and Library PreparationFollowing the capture of RNA from individual cells, reverse transcription and amplification are performed to generate sufficient cDNA for downstream sequencing analysis [28]. Several reverse transcription initiation strategies are commonly employed, including the Poly(A) Tailing method, the Template-Switching (TS) approach, and random primer-based methods designed for non-poly(A) RNA capture (e.g., SUPeR-seq, snHH-seq, and snRandom-seq) [31-34]. Subsequently, the cDNA is subjected to pre-amplification, during which Multiple Annealing and Tailing-based Quantitative Single-cell RNA-seq (MATQ-seq) utilizes a multiplex annealing and tailing strategy to substantially reduce amplification noise, thereby improving sensitivity and quantitative accuracy [35]. The integration of barcodes and Unique Molecular Identifiers (UMIs) before or during amplification (e.g., Single-Cell RNA Barcoding and sequencing (SCRB-seq), MULTIplexing using lipid-Tagged Indices for single-cell RNA sequencing (MULTI-seq)) allows for the correction of quantification errors caused by PCR duplicates [18,36]. Subsequently, the amplified cDNA is subjected to a series of processing steps, including fragmentation, end repair and tailing, adapter ligation, and library PCR enrichment, ultimately yielding libraries customized for specific research objectives.
Post-High-Throughput Sequencing Data Processing and AnalysisSingle-cell libraries are sequenced using high-throughput sequencing platforms (short-read platforms: Illumina, Ion Torrent, MGI; long-read platforms: PacBio, Oxford Nanopore) to generate raw sequencing data (reads) [37-42]. Raw data files generated by sequencing instruments are converted into a standardized FASTQ format. In the context of data quality control, tools such as FastQC are used to evaluate the quality of raw FASTQ files, focusing on metrics such as base quality distribution, sequence length distribution, Guanine-Cytosine content, and adapter contamination. Subsequently, Trimmomatic or Cutadapt is employed to remove low-quality bases (“quality trimming”) and adapter sequences (“adapter trimming”). Quality control reports generated by these tools, along with those from downstream processes, are consolidated using MultiQC [19,43-44]. Clean reads are aligned to the reference genome using STAR, Kallisto, or Cell Ranger to generate a cell-by-gene expression matrix containing gene’s UMI counts. Quality control and filtering of the expression matrix (Expression Matrix QC) are performed to remove abnormal cells based on predefined thresholds. Background contamination was subsequently corrected using SoupX to adjust for ambient RNA, and low-abundance genes expressed in fewer than a certain number of cells [45].
In the context of data preprocessing, normalization procedures are applied to mitigate technical biases such as differences in sequencing depth and cell capture efficiency, thereby ensuring comparability of gene expression levels across distinct cell populations. Specifically, the scran package (used for estimating cell size factors) and the Seurat::NormalizeData/SCTransform methods were utilized to remove variations in sequencing depth [46]. For the selection of highly variable genes, informative genes were identified using Seurat::FindVariableFeatures or scran::modelGeneVar. Subsequently, Seurat::ScaleData (centering and scaling) was applied to the highly variable genes for normalization. After gene normalization, Principal Component Analysis (PCA)-based dimensionality reduction was performed. Batch effects were then integrated into the PCA space (as opposed to the gene expression matrix) using Harmony or MNNCorrect. The preprocessed data are saved as Seurat or AnnData objects and stored in universal file formats, such as h5ad, for downstream analysis, which can include cellular clustering and subpopulation identification, differential expression analysis, data visualization, and interpretation [47,48].
Application of Single-Cell Sequencing Technology in Influenza Virus Infection ResearchSCS technology has been widely applied to investigate cellular heterogeneity profiles following infection with the influenza virus. Cells were categorized based on immune function into immune and non-immune cells, and further classified according to infection status into infected cells, bystander cells, and uninfected cells. Cluster analysis has identified key cellular subpopulations that, based on differentially expressed genes, provide novel insights into the interactions between influenza viruses and various cell types. A systematic review of recent applications of SCS technology in influenza virus research is shown (Figure 2 and Table 1).
Figure 2. illustrates that research on the SCS technology in influenza viruses primarily centers on analyzing differentially expressed genes (DEG) across various cell types and characterizing immune cell differentiation following infection. (Created with BioGDP.com)
Table 1. Applications, advantages, and limitations of different single-cell sequencing technologies in influenza research
| technology | Influenza research | advantage | boundedness |
|---|---|---|---|
| scRNA-seq | Differences in viral RNA load and distribution across cell types [49]; Key cellular sources of inflammatory factors [50]; Cellular heterogeneity between infected and bystander cells [51]; Comparative immune characteristics of cross-species infections [52-53]; Comparative analysis of IAV and COVID-19 infections, including co-infection with Streptococcus pneumoniae [54-57]; Macrophage polarization and its association with viral susceptibility [58]; Peripheral immune profiles across infected populations [59]; Development of porcine animal models [60]; Lymphatic system remodeling during infection [61]; Impact of environmental humidity on infection dynamics [62]; Memory CD8+ T cell responses following primary infection and reinfection [63]; Differentiation trajectories of plasmacytoid dendritic cells (pDCs) [64]; Vaccine-induced transcriptional responses, dynamic shifts in immune cell subsets, and adjuvant research [65-66] | High-throughput, capable of revealing cellular heterogeneity and enabling simultaneous analysis of host and polyA-tailed viral transcriptomes | Viral transcripts exhibit low expression levels, the method is unsuitable for analyzing viral RNAs lacking polyA tails, and technical noise is substantial. |
| Combined with spatial transcriptomics, this study aims to elucidate spatial differences in the lungs of young versus old mice following infection, as well as the influence and underlying mechanisms by which age affects B cell differentiation trajectories [67] | Preserves spatial information and enables direct correlation between infected areas and the immune microenvironment | The resolution fails to achieve true single-cell level, as most spots contain multiple cells; the cost is prohibitively high, and data analysis is complex | |
| scBCR-seq/scTCR-seq | Transcriptional differences and high diversity of memory B cells in the lungs and lymphoid organs [68]; Functional regulation of tissue-resident memory B and T cells [115-116, 122] | High-throughput analysis of adaptive immune responses, discovery of neutralizing antibody clones, and elucidation of immune memory mechanisms | It is primarily limited to lymphocytes and cannot directly provide full transcriptome information, often requiring combination with single-cell RNA sequencing (scRNA-seq) |
| mudRapp-seq | Interconnections between viral RNA (vRNA and mRNA) and cellular heterogeneity [69] | Direct RNA detection without reverse transcription, high sensitivity, high specificity, and preservation of spatial information | There are relatively few application cases, probe design is complex, and the approach requires dedicated instruments and specific operational procedures |
| scATAC-seq | The epigenomic and transcriptional landscapes of human immunity to seasonal and pandemic influenza vaccines [70] | Uncover the upstream regulatory mechanisms—such as transcription factor activity—that drive transcriptional heterogeneity; integrate with single-cell RNA sequencing (scRNA-seq) to construct a comprehensive gene regulatory network | Direct detection of virus-related changes is not feasible, the data are sparse and analysis is complex, and integration with single-cell RNA sequencing (scRNA-seq) is required to obtain a comprehensive understanding |
Cellular Heterogeneity During Viral Infection
Heterogeneous Distribution of Influenza Viral mRNAThe replication of the influenza virus within host cells involves multiple stages, during which genomic replication triggers a series of changes in host cell gene expression [71]. Upon infecting cells or in mice, the influenza virus exploits host factors and proteins to support its replication and proliferation. By integrating UMI and cellular barcode information, the dynamic abundance of influenza viral mRNA within cells can be accurately quantified [72,73]. Following viral infection of A549 cells, most cells exhibited minimal or undetectable levels of viral mRNA. Over time, progressive accumulation of viral mRNA was observed in the infected cells. Analysis of viral mRNA dynamics indicated the activation of viral clearance mechanisms approximately one week post-infection [49,50]. Analysis of viral mRNA abundance in the lung tissues of infected mice revealed a higher proportion of infected cells, with epithelial cells exhibiting the highest infection rate and T cells the lowest. Additionally, the infection rate of non-immune cells was higher than that of immune cells [51]. Compared to A549 cells, the pulmonary microenvironment in murine lung tissue is more complex, especially with regard to tissue specificity and cellular interactions, highlighting the need for more in-depth investigations [74]. Additionally, studies have identified an intriguing phenomenon in which most infected cells show minimal viral mRNA presence, whereas a minority of infected cells exhibit viral mRNA comprising over half of the cellular transcriptome [49,75]. In-depth analysis suggests two potential explanations: first, the absence or varying expression ratios of viral genomic mRNA fragments may contribute to the elevated viral mRNA abundance observed in a subset of cells [49,58]. For instance, during influenza virus infection, viral replication shifts from the transcription phase to the replication phase. The NS2 protein, efficiently synthesized through early transcription, interacts with the RNA-dependent RNA polymerase (RdRp) to facilitate the transition of viral RNA from transcription to replication. When NS2 protein expression decreases, viral replication levels decline. Cell-to-cell variations in NS2 expression can lead to differences in viral mRNA abundance [76-79]. Second, this phenomenon may also be attributed to variations in the number of cellular receptors, as the distribution of sialic acid receptors is heterogeneous across different cell types, and even within the same cell type, heterogeneity exists depending on differentiation status or cell cycle phase [80]. The study conducted by Ni Z et al. showed that after the viral hemagglutinin (HA) protein binds to metabotropic glutamate receptor 2 (mGluR2), mGluR2 interacts with the calcium-activated potassium channel (KCa1.1), thereby participating in the initiation and completion of clathrin-mediated endocytosis (CME) of influenza viruses. The expression level of mGluR2 directly influences the efficiency of influenza virus cellular entry. Variations in viral mRNA content across cells may correlate with differences in mGluR2 expression levels in those cells [81]. The extreme heterogeneity in viral mRNA distribution not only reflects differences in replication efficiency but also shows a significant association with host cell cycle regulation (e.g., downregulation of G2-M checkpoint genes) and oxidative stress responses (e.g., activation of the Nrf2 pathway). These findings suggest that the virus may exploit host transcriptome remodeling as a mechanism for immune evasion. Recent technological advancements highlight the potential of mudRapp-seq in elucidating viral replication mechanisms. By analyzing the heterogeneity of viral mRNA abundance across different cell types, this approach deepens our understanding of viral infections [69].
Dynamics of Pro-inflammatory Factors after Influenza InfectionSingle-cell analysis of DEGs revealed significant upregulation of antiviral-related signaling pathways in infected cells, such as the IFN and IRF7 signaling pathways. Simultaneously, the principal components involved in antiproliferative and inflammatory processes is also elevated. The expression levels of key transcriptional regulators of host immune responses, such as STAT3, NFKB1, and REL, are upregulated. Additionally, several cytokines with incompletely characterized functions in influenza virus infection, including CHD1, BCLAF1, and PHF3, also show increased expression [51,56,59-60]. Each immune cell subset plays distinct or overlapping roles and engages in intercellular interactions. The antiviral effects are evident across consistent cell types—including NK cells, B cells, T cells, and neutrophils—irrespective of their specimen source. This was demonstrated in samples from PR8 H1N1-infected mice (lungs and spleens), individuals with confirmed IAV infection (peripheral whole blood from children, adults, and pregnant women), and children with severe H5N6 avian influenza (PBMC samples). Although immune responses induced by different influenza viruses are broadly similar, infection with the H5N6 avian influenza virus tends to be more severe [50-51,53,59,67-68]. The research team provided an overview of the current key findings (Table S1), with specific emphasis on pro-inflammatory factors. Neutrophils are a key cell type involved in influenza infection [56,57]. Zhang et al. performed an analysis demonstrating that the release of pro-inflammatory factors induced by influenza virus infection occurs in two distinct phases [50]. Kasmani MY et al. propose that the incidence of pulmonary inflammation rises with advancing age, and by applying spatial sequencing technology and scRNA-seq data, they have identify temporal and age-associated changes in neutrophil populations [67]. Initial clustering and Gene Ontology (GO) enrichment analyses were performed, followed by the identification of cell types expressing pro-inflammatory factors based on single-cell-specific marker genes. PD-L1-high neutrophils were identified as the primary contributors to the first wave of proinflammatory factor release. Subsequent transcriptional profiling of inflammatory responses in the second wave of cell populations, combined with Pearson correlation analysis, revealed that Pf4-high macrophages are the main source of the second wave of pro-inflammatory factors [50]. In young mice post-infection, the expression levels of neutrophil inflammatory regulatory genes, including IL-1α, CCL3, CXCL3, and CXCL1, were elevated, with CXCL chemokine signaling notably exceeding the release levels observed in neutrophils from aged mice. Single-cell DGE data revealed a contrasting pattern compared to the generally more severe inflammatory condition observed in older mice. Some studies attribute this discrepancy to the masking effect of neutrophil quantity on functional quality. Kulkarni U et al. confirmed that aged mice exhibit higher neutrophil counts following influenza infection [67,82]. The analysis of pro-inflammatory factors should be expanded to include the inflammatory regulatory network to provide a more comprehensive understanding of the host response following infection. Single-cell differential gene expression DGE data can be employed to examine mitochondrial-related and coagulation-associated genes. Furthermore, such data also enables the comparison of how varying humidity levels affect influenza virus infection [62,67]. The aforementioned analysis primarily aims to elucidate the origins of pro-inflammatory factors during influenza infection, thereby enabling a more comprehensive integration of both global and local perspectives in the conceptualization of the infection process.
Dynamic Profiling of Host Immune Cell Populations During InfectionThe cellular heterogeneity of virus-infected cells is reflected in the dynamic responses of host immune cells. Following viral infection, this heterogeneity among immune cell populations is predominantly characterized by the activation or suppression of specific signaling pathways, which arise from differential gene expression across distinct subpopulations or are modulated by various regulatory factors.The differentiation patterns of various immune cell types following infection, as well as the interrelationships among their respective subsets, are analyzed using single-cell sequencing of immune cells. Future investigations should focus on exploring the cellular heterogeneity of B and T cells in response to influenza virus infection.
Heterogeneity and dynamic regulation of T-cell subsetsFollowing the presentation of viral antigens by dendritic cells (DCs), CD8+ T cells are activated and differentiate into cytotoxic effector T cells (CTLs). These CTLs mediate their cytotoxic effects through two distinct mechanisms: direct cytolysis of infected cells via the granzyme/perforin pathway or induction of target cell apoptosis through the Fas/FasL signaling pathway [83-85]. They secrete IFN-γ to inhibit viral replication and mediate antiviral and immunomodulatory effects through cytokines, such as TNF-α [86,87]. CD4+ T cells primarily differentiate into Th1 and Tfh subsets and can also give rise to other subsets, such as Th17 cells, which play a critical role in the immune response to influenza virus infection [88]. CD4+ T cells play a central role in enhancing the functionality of CTLs, promoting antibody production by B cells, and supporting mucosal defense mechanisms [89]. Following pathogen clearance, a subset of effector T cells differentiates into heterogeneous memory T cell populations, including central memory T cells (Tcm) located in lymphoid tissues, stem cell-like memory T cells (Tscm), effector memory T cells (Tem) found in peripheral tissues, and tissue-resident memory T cells (Trm) that permanently reside in respiratory mucosal tissues [90,91]. Memory cells can rapidly respond to reinfection at local sites and efficiently migrate to infection foci via circulatory pathways, thereby mediating immunological functions [92]. Regulatory T cells (Tregs) play a critical role in modulating excessive immune responses and maintaining immune homeostasis during influenza virus infections [93]. During influenza virus infection, T cells may undergo exhaustion, characterized by functional impairment, which can impair viral clearance and delay disease recovery [94,95].
SCS technology facilitates a more comprehensive exploration of underlying mechanisms. Through clustering and visualization analysis, multiple CD4+ and CD8+ T cell subsets can be identified, which are significantly enriched in biological processes associated with oxidative stress and cell death [53,67]. Furthermore, the analysis of CD4+T cell subsets can reveal the regulatory factors governing each subset through pseudotime trajectory analysis, thereby identifying the subsets involved in immune and inflammatory pathways [53]. Similar methodologies have been applied in studies of memory CD8+T cells, where tissue-resident memory T cells (CD8+Trm) accumulate in the lungs following infection and concurrently exhibit sustained high expression of CD49a for up to 90 days. The upregulated DEGs in CD8+ Trm cells are predominantly enriched in the FoxO signaling pathway, apoptosis, PD-L1 expression, PD-1 checkpoint pathway, and adherens junctions. According to the KEGG database. Following reinfection, both effector memory T cells (CD8+ Tem) and CD8+Trm cells show enrichment in the PI3K-Akt-mTOR and type I interferon signaling pathways [63]. In T cell research, it is essential to obtain comprehensive data that elucidate the cellular heterogeneity of naïve, effector, and memory T cells following influenza virus infection. Such data would significantly contribute to drug development and deepen our understanding of immune mechanisms.
Organ-specific differentiation of B cells during influenza infectionIn influenza virus infection, B cells play a pivotal role through a multi-stage differentiation process. Initially, their surface B cell receptors (BCRs) specifically recognize viral antigens, such as HA, and are activated with T cell help. Subsequently, they enter the germinal center (GC), where they undergo somatic hypermutation (SHM) and affinity maturation. This process involves the random introduction of BCR mutations via activation-induced cytidine deaminase (AID), ultimately leading to the selection of high-affinity clones [96-99]. Simultaneously, class switch recombination (CSR) leads to the production of IgG or IgA antibodies. Mucosal IgA antibodies can prevent viral invasion of epithelial cells, whereas IgG antibodies exert their effects by neutralize the virus or recruit immune effector cells [100,101]. The integration of scRNA-seq and scBCR-seq allows for the precise characterization of B cell immune responses and the functional analysis of memory B cells following influenza virus infection. Using the Immcantation pipeline and scRepertoire, BCR sequence data can be systematically integrated into comprehensive multi-omics analyses [102-104]. Cell clustering analysis based on genes associated with specific differentiation states revealed that genes related to IgA antibody secretion and B cell receptor expression were significantly enriched in HA-specific memory B cells (HA-Bmems) and plasma blasts (PBs) across all examined organs. Notably, IgA-secreting cells showed preferential enrichment in these two cell populations. HA-positive B cell clusters display a high degree of organ specificity and lack temporal specificity [68]. To decipher the single-cell-level differentiation patterns of memory B cells (Bmems), trajectory analysis was performed using Slingshot, and RNA velocity analysis was performed with scVelo, revealing the distinct transcriptional profile of pulmonary Bmems marked by activation and tissue-residency features [68,105-106]. Distinct microenvironments provided by different tissues and organs promote B cell differentiation. These analytical findings demonstrate a high degree of inter-organ dissemination of GC-derived HA-Bmems [68].
In addition to B and T cells, the cellular heterogeneity of other cell types following influenza virus infection highlights their influence on the infection microenvironment. As key components of the innate immune system, macrophages display diverse immune functions and are defined by distinct polarization states, underscoring their complex heterogeneity [107,108]. Yu et al. elucidated the influence of macrophages with distinct polarization states on T cell responses following influenza infection; utilizing time-resolved single-cell sequencing and metabolic RNA labeling techniques [58]. Influenza infection triggers a substantial increase in pulmonary lymphatic vessel density, accompanied by extensive proliferation of lymphatic endothelial cells (LECs) in the lungs, and a novel PD-L1-expressing subpopulation was identified that persists during viral infection and suppresses LEC differentiation and/or proliferation [61,109]. Plasmacytoid dendritic cells (pDCs), a rare subset of the innate immune system, have a distinctive ability to produce large amounts of type I interferons. During influenza virus infection, these cells shift from being specialized cytokine hyperproducers to adopting antigen-presenting cell (APC)-like features, thereby exhibiting transcriptional diversity [64,110-111]. The study of cellular heterogeneity, enabled by the integration of multiple advanced technologies, has substantially advanced our understanding of host cell responses to influenza virus infection.
The impact of aging on T and B cell susceptibility to influenza virus infectionAging profoundly reshapes the host's immune response to influenza viruses through mechanisms of immunosenescence and chronic inflammation [112]. In the rhesus monkey model, the numbers of alveolar macrophages and infiltrating macrophages were significantly increased in the elderly group, while the number of T cells decreased concurrently [113]. The major coding genes differ between the young and the elderly. Aging leads to dysfunction of key cytolytic and memory functions in T cells, and the expression of multiple T-cell exhaustion markers shows an upward trend [67]. Wang et al. discovered in children that a subset of B cells exhibits a potentially protective cytotoxic effect, which diminishes with age [114]. The differentiation of B cells following influenza virus infection is influenced by age. Elderly individuals' B cells tend to differentiate into plasma cells rather than memory cells, which contributes to reduced vaccine efficacy [67]. Alice R. Burton et al. discovered that hemagglutinin-specific memory B cells formed in young individuals exhibit an FcRL5+ atypical phenotype, potentially originating from pre-GC precursor cells, and show evidence of somatic hypermutation and positive selection. In contrast, these features are less pronounced in the elderly population, confirming impairments in the germinal center reaction and memory B cell response following vaccination [115]. After vaccination, young individuals exhibit a stronger clonal response compared to the elderly. The proportion of plasmablasts is reduced in older adults. Differential abundance analysis has also identified a greater number of vaccine-responsive cells that do not participate in expanded clones, a feature particularly prominent in the elderly population [116]. These findings not only elucidate the mechanisms underlying the heightened susceptibility to influenza and reduced vaccine efficacy in the elderly, but also provide critical scientific foundations for developing novel adjuvants and immune-targeted interventions for this population, as well as for the advancement of universal vaccine research.
Molecular Mechanisms Underlying Vaccine DevelopmentThe development of influenza vaccines has continually faced several major challenges: the rapid mutation of influenza viruses via antigenic drift and antigenic shift necessitates annual vaccine updates and hinders the attainment of broad-spectrum efficacy, the existence of substantial variations in vaccine-induced immune responses across different population groups, and ongoing evaluations of the safety profile of live attenuated vaccines [117-121]. SCS technology enables researchers to construct a comprehensive immunome atlas at single-cell resolution following influenza vaccination, thereby facilitating in-depth analysis of cellular heterogeneity and molecular regulatory networks in immune responses.The integration of multiple technologies and methodologies has substantially advanced influenza vaccine research. The authors summarize the common applications of scRNA-seq in vaccine studies (Table 2), offering novel insights into vaccine-induced innate immune training, lymphocyte differentiation, and the formation of tissue-resident memory T cells.
Table 2. The combined application of single-cell RNA sequencing (scRNA-seq) with multiple technologies in influenza vaccine research
| technical combination | research problem | main discovery |
|---|---|---|
| scBCR-seq | Vaccines exhibit a relatively weaker protective effect in the elderly, and age may influence the quantity or type of B cells | The immune response following vaccination declines with age, and the magnitude of plasmablast expansion is greater in younger individuals than in older adults [116] |
| Characteristics of B cell specificity, function, and subsets induced by chimeric hemagglutinin (cHA) vaccines | The cHA vaccine enriches stem-binding B cells within the memory B cell compartment one year after vaccination [122] | |
| EpiTOF, scATAC-seq | At the single-cell level, there remains a gap in the comprehensive map of the human epigenome during the immune response process | After influenza vaccination, chromatin accessibility at the AP-1 site in myeloid cells decreases; the AS03 adjuvant enhances accessibility in the IRF/STAT binding region [70] |
| scRNA-seq + Indexed Flow Sorting (IFS), scBCR-seq | How does aging impact the memory B cell response | After vaccination, the elderly exhibit deficiencies in both the germinal center (GC) response and the memory B cell response [115] |
| scBCR-seq, Fluorescence Activated Cell Sorting (FACS) | The cell types responsible for IgA production in the lower respiratory tract following intranasal vaccination remain unclear | This study investigated the phenotypic characteristics, residency status, and functional roles of IgA-secreting B cells in the lungs and confirmed that the development of these cells depends on CXCR3 signaling [100] |
| Protein Microarray (PM) | Explore the mechanism by which adjuvants enhance the breadth of cross-reactivity | Adjuvants enhance the magnitude and durability of the antibody response to vaccines [65] |
| Longitudinal Antibody Repertoire Sequencing (LAR-Seq) | Analyze the molecular and cellular characteristics of the antibody response following influenza vaccination | Numerous antibody clones induced by vaccination do not bind to the vaccine antigen and instead activate non-specific bystander antibodies through the bystander effect [66] |
One week after vaccination, a significant increase in the relative proportion of plasma cells was observed, which was associated with the production of protective neutralizing antibodies. Monocytes, DCs, CD8+T cells, natural killer (NK) cells, and γδ T cells showed a decreasing trend, all of which recovered by the second week, revealing dynamic changes in immune cell populations following vaccination [123]. Following vaccination, transcriptomic alterations were observed across various immune cell populations, displaying distinct patterns dependent on cell type specificity. These changes are primarily associated with activation processes, clonal expansion induction, and antiviral response mechanisms [123,124]. Research on the safety of influenza vaccination warrants attention. A comprehensive safety assessment must consider the immune status and underlying conditions of diverse populations. At the population level, inactivated influenza vaccines are generally safe for older adults and immunocompromised individuals. However, immunosenescence in the former may slightly increase the risk of non-specific adverse events such as fever and fatigue, while the latter requires long-term monitoring due to potential risks associated with immune activation [125-127]. For pregnant women, inactivated vaccines serve as a crucial protective measure and have been proven safe throughout all stages of pregnancy [128]. At the technical level, the type of vaccine directly determines differences in risks such as inflammation and thrombosis [129]. Research on molecular mechanisms provides a profound explanation for this: inactivated vaccines do not upregulate platelet aggregation or pro-inflammatory genes, confirming their safety advantages. In contrast, adenovirus-vector vaccines, which mimic natural infections, generate a distinct pulmonary inflammatory environment. Understanding these differences is crucial for vaccine design [123,130]. Safety serves as the cornerstone of vaccine development. On this foundation, novel vaccines—such as cHA vaccines—designed to elicit broad-spectrum and long-lasting immune responses, together with strategies combining adjuvants like AS03, are advancing into a new stage of enhancing the balance between immunity and tolerance through coordinated epigenomic regulation [70,122]. Intranasal vaccines display distinct characteristics compared to other vaccine types, as they promote the secretion of IgA antibodies [131].To elucidate the origin of IgA in the pulmonary cavity, the phenotype, residency, and function of IgA-secreting B cells in the lung were analyzed using scBCR-seq, confirming that tissue-resident memory B cells are the primary source of IgA [100]. Simultaneously, a rare subpopulation was identified among keratinocyte nasal immune interaction front epithelium (KNIIFE) cells, which showed a concurrent increase in tissue-resident memory T-like cells. The presence of the CXCL16-CXCR6 axis in these populations has substantially contributed to the comprehensive mapping of nasal infection dynamics [132]. Adjuvants have been shown to enhance vaccine immunogenicity. However, in a comparative study between the self-amplifying mRNA vaccine (SAM-H1/CNE) for influenza A (H1N1) virus and the MF59-adjuvanted monovalent influenza vaccine, the SAM-based vaccine demonstrated superior efficacy in inducing stronger and more robust CD8+ T cell responses [133-135]. The first investigation of influenza vaccines utilizing SCS technology was conducted in a llama model, providing a unique perspective for vaccine and antibody development, thus advancing innovative research in this field [136]. Based on single-cell sequencing data, researchers have developed a vaccine response prediction model, highlighting the urgent need for integrating additional predictive models and artificial intelligence technologies [137]. The safety and efficacy of influenza vaccines remain central research priorities. Heterogeneous cellular data have provided new insights into this field, laying a critical foundation for the development of next-generation vaccines.
Conclusion and Future Directions
The SCS technology has significantly advanced influenza virus research by leveraging the heterogeneous responses of distinct cellular subpopulations to overcome these limitations. This approach has enabled a qualitative leap in our understanding of immune cell population differentiation and dynamics following infection.Through the analysis of single-cell differential expression profiles, researchers have gained deeper insights into the regulatory networks activated after viral infection. Furthermore, the establishment of a specialized single-cell database for influenza viruses is now feasible. Such a database would enable refined analysis of host cell subpopulations by integrating multi-timepoint and multi-tissue data, for instance, from the spleen and lung. Additionally, a cross-species single-cell database encompassing avian, swine, and human hosts should be developed to identify key cellular targets involved in viral cross-species transmission. Moreover, the dynamic expansion patterns of virus-specific T/B cell clones can be systematically monitored. To realize this vision and enhance its scientific value, a standardized pipeline has been implemented for rigorous quality control, batch correction, and automated cell annotation to ensure data comparability and accuracy. A model combining annual major releases with quarterly incremental updates is recommended, along with the sharing of pre-trained analysis models by referencing the scvi-hub framework. In this way, the database can evolve from a static archive into an intelligent platform capable of continuously supporting dynamic analysis and hypothesis generation [138]. Based on the single-cell characteristics of lung tissues from severe influenza patients, integrated with metabolic differences in host genes such as CES derived from drug-sensitive and drug-resistant patient data, this study adopts a multidimensional and multi-perspective approach to offer systematically oriented strategic insights into the infection mechanisms and therapeutic strategies of the influenza virus [139]. In the field of vaccine and drug development, this single-cell database facilitates the elucidation of B cell cross-reactivity to conserved epitopes within the HA stem region, reveals the differential activation mechanisms of adjuvants across dendritic cell subsets (cDC1/cDC2), and offers a comprehensive understanding of the regulatory networks through which viruses hijack host metabolic pathways, as revealed by multi-omics integrated sequencing [140-142]. Consequently, enhancing the depth and breadth of existing databases is essential. The development of these databases requires standardized frameworks to improve comparability, and dynamic updates to continuously incorporate data on emerging strains. Furthermore, fostering interdisciplinary collaboration across fields, such as virology, computational biology, and clinical medicine, is essential to enhance the utility and coverage of databases. The integration of artificial intelligence, particularly through deep learning approaches, to predict virus-host interaction networks will significantly accelerate the development of anti-influenza strategies [143].
Multi-omics integration represents a key strength and an emerging direction in SCS technology. By leveraging bioinformatics analysis to capture cellular heterogeneity, it can be synergistically integrated with spatial transcriptomics, immune cell sorting, mass spectrometry, and other advanced techniques to generate comprehensive datasets; thereby enabling more precise and in-depth research outcomes [144]. For instance, spatial transcriptomics technologies (e.g., 10x Visium and Slide-seq) preserve the spatial localization of cells within tissues, thereby elucidating the spatial distribution of distinct immune cell subpopulations. The integration of spatiotemporal information enables the reconstruction of spatiotemporal trajectories during cell differentiation. The combined use of spatial transcriptomics and single-cell sequencing compensates for the loss of in situ tissue information; thereby resolving the spatial distribution of virus-infected cells and immune cell infiltration patterns in lung tissue [145-148]. The integration of multi-omics analysis with single-cell sequencing technologies enables a comprehensive understanding of infection-induced immune regulatory mechanisms and the identification of precise therapeutic targets. This includes epigenomic profiling (single-cell ATAC-seq for chromatin accessibility, scCOOL-seq for chromatin state and DNA methylation), proteomic analysis (CITE-seq for surface protein detection and CyTOF), and metabolomic characterization (single-cell metabolic mass spectrometry imaging). When combined with in vivo dynamic tracking and lineage tracing technologies (CRISPR barcoding, fluorescent reporter systems), these omics approaches, together with SCS technology, facilitate in-depth analysis of dynamic epigenetic modifications in memory cells following influenza virus infection, thereby identifying key molecular determinants of cell fate [149-156]. Furthermore, the integration of computational biology with artificial intelligence can accelerate the development of SCS technologies, facilitating large-scale data mining, cross-species comparisons, and the organization of clinical information, among other applications[157].
Certainly, there is still room for improvement in Single-Cell SCS technology. For instance, the preparation of single-cell suspensions may result in the loss of microenvironmental information from tissues. Given the low abundance of influenza virus mRNA in most infected cells, single-cell sequencing may fail to detect certain viral signals. Additionally, low-abundance viral genes might be masked by highly expressed host genes (e.g., interference from mitochondrial transcripts) [158,159]. In the future, there is a need to develop virus-specific primer enrichment technologies or to optimize data analysis algorithms, constructing a comprehensive training dataset integrating multi-dimensional features such as sequence k-mer frequency, alignment quality scores (e.g., MAPQ), coverage depth uniformity, and sequence context embedding; Employ convolutional neural networks (CNNs) or Transformer models to automatically learn deep sequence patterns and features within viral genomes. By incorporating weighted loss functions and transfer learning strategies, effectively address the challenges of extreme class imbalance—where viral reads constitute an extremely low proportion—and sparse annotated data [160-162]. Furthermore, developing conserved sequence capture probes for influenza viruses is feasible, and their integration with single-cell RNA sequencing can significantly enhance the sensitivity of viral gene detection. Alternatively, utilizing PacBio or Oxford Nanopore Technologies (ONT) platforms enables the direct capture of complete viral genomes, thereby facilitating a comprehensive analysis of quasispecies diversity [163]. Single time point sequencing is unable to capture dynamic processes; therefore, it is critical to employ tools such as Monocle3 to construct infection progression models that simulate the continuum from viral entry and replication to host cell apoptosis [164]. Furthermore, developing advanced tools such as Viral-Track is essential to automatically segregate host and viral reads, enabling simultaneous analysis of host gene expression and viral genomic variations. This approach overcomes the limitations of existing algorithms, which are predominantly designed for single-species analysis [165]. Although single-cell sequencing technology has become a powerful tool for influenza virus research, its high sequencing costs and complex data analysis workflows remain barriers to its widespread adoption in certain studies.Future efforts should focus on refining technical protocols, reducing sequencing costs, and developing more efficient bioinformatics tools to fully harness the potential of this technology. This study presents the first systematic integration of single-cell sequencing in three-dimensional applications, including investigations into influenza virus infection mechanisms, immune cell dynamics, and vaccine development. It highlights the pivotal role of technological convergence in advancing future research, aiming to provide novel insights for the development of precise antiviral strategies.
Abbreviations
Activation-induced cytidine deaminase: AID; Antigen-present-ing cell: APC; Assay for Transposase-Accessible Chromatin with sequencing: ATAC-seq; B cell receptors : BCRs; Cellular Indexing of Transcriptomes and Epitopes by sequencing: CITE-seq; Clathrin-mediated endocytosis: CME; Convolutional neural networks: CNNs; Clustered Regularly Interspaced Short Palin-dromic Repeats: CRISPR; Class switch recombination: CSR; Cy-totoxic effector T cells: CTLs; Cytometry by Time-Of-Flight: Cy-TOF; Dendritic cells: DCs; Differentially expressed genes: DEG; Droplet-based single-cell RNA sequencing: Drop-seq; Fluores-cence-Activated Cell Sorting: FACS; Germinal center: GC; Gene Ontology: GO; Hemagglutinin: HA; Confirmed influenza A virus: IAV; Indexed Flow Sorting: IFS; Keratinocyte nasal immune interaction front epithelium: KNIIFE; Longitudinal Antibody Repertoire Sequencing: LAR-Seq; Lymphatic endothelial cells: LECs; Multiple Annealing and Tailing-based Quantitative Sin-gle-cell RNA-seq: MATQ-seq; Metabotropic glutamate receptor 2: mGluR2; MULTIplexing using lipid-Tagged Indices for sin-gle-cell RNA sequencing: MULTI-seq; Natural killer: NK; Oxford Nanopore Technologies: ONT; Peripheral Blood Mononuclear Cell: PBMC; Principal Component Analysis: PCA; Plasmacy-toid dendritic cells: pDCs; Protein Microarray: PM; RNA-de-pendent RNA polymerase: RdRp; Single-cell B-cell receptor/T-cell receptor sequencing: scBCR-seq/scTCR-seq; Single-Cell RNA Barcoding and sequencing: SCRB-seq; Single-cell RNA sequencing: scRNA-seq; Single-cell sequencing: SCS; Somatic hypermutation: SHM; high-throughput and high-sensitivity sin-gle-nucleus total RNA sequencing: snHH-seq; Single nucleus Random-seq: snRandom-seq; Single-cell Universal Poly(A)-in-dependent RNA sequencing: SUPeR-seq; Central memory T cells: Tcm; Effector memory T cells: Tem; Regulatory T cells: Tregs; Tissue-resident memory T cells: Trm; Template Switch-ing: TS; Stem cell-like memory T cells: Tscm; Unique Molecular Identifiers: UMIs.
Supplementary Materials
Declarations
Author Contributions
Xingting Li : Data curation, Formal analysis, Writing : original draft; Lingxi Gao : Conceptualization, Methodology, Writing : review & editing, Supervision. All authors read and approved the final manuscript.
Acknowledgments
The authors thank the colleagues and students from the De-partment of Microbiology for their valuable discussions, and specifically acknowledge the BioGDP platform for providing professional illustration services for this article.
Funding Information
This work was supported by the Natural Science Foundation of Guangxi (2024GXNSFAA010427).
Ethics Approval and Consent to Participate
Not Applicable.
Competing Interests
The authors declare that they have no existing or potential commercial or financial relationships that could create a con-flict of interest at the time of conducting this study.
Data Availability
Not Applicable (this is a review article and no new data were generated).
References
 Figures
Figures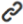 References
References Peer
Peer Information
InformationFigure 1. The experimental workflow of scRNA-seq technology involves single-cell isolation and capture, followed by nucleic acid amplification and library construction, selection of an appropriate high-throughput sequencing platform, and concludes with data processing and bioinformatics analysis.
Figure 2. illustrates that research on the SCS technology in influenza viruses primarily centers on analyzing differentially expressed genes (DEG) across various cell types and characterizing immune cell differentiation following infection. (Created with BioGDP.com)
Peer-review Terminology
Identity transparency: Single anonymized
Reviewer interacts with: Editor
Details
This is an open access article under the terms of the Creative Commons Attribution License(http://creativecommons.org/licenses/by/4.0/), which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Publication History
Received 2025-07-15
Accepted 2025-10-12
Published 2025-12-14