




Abstract
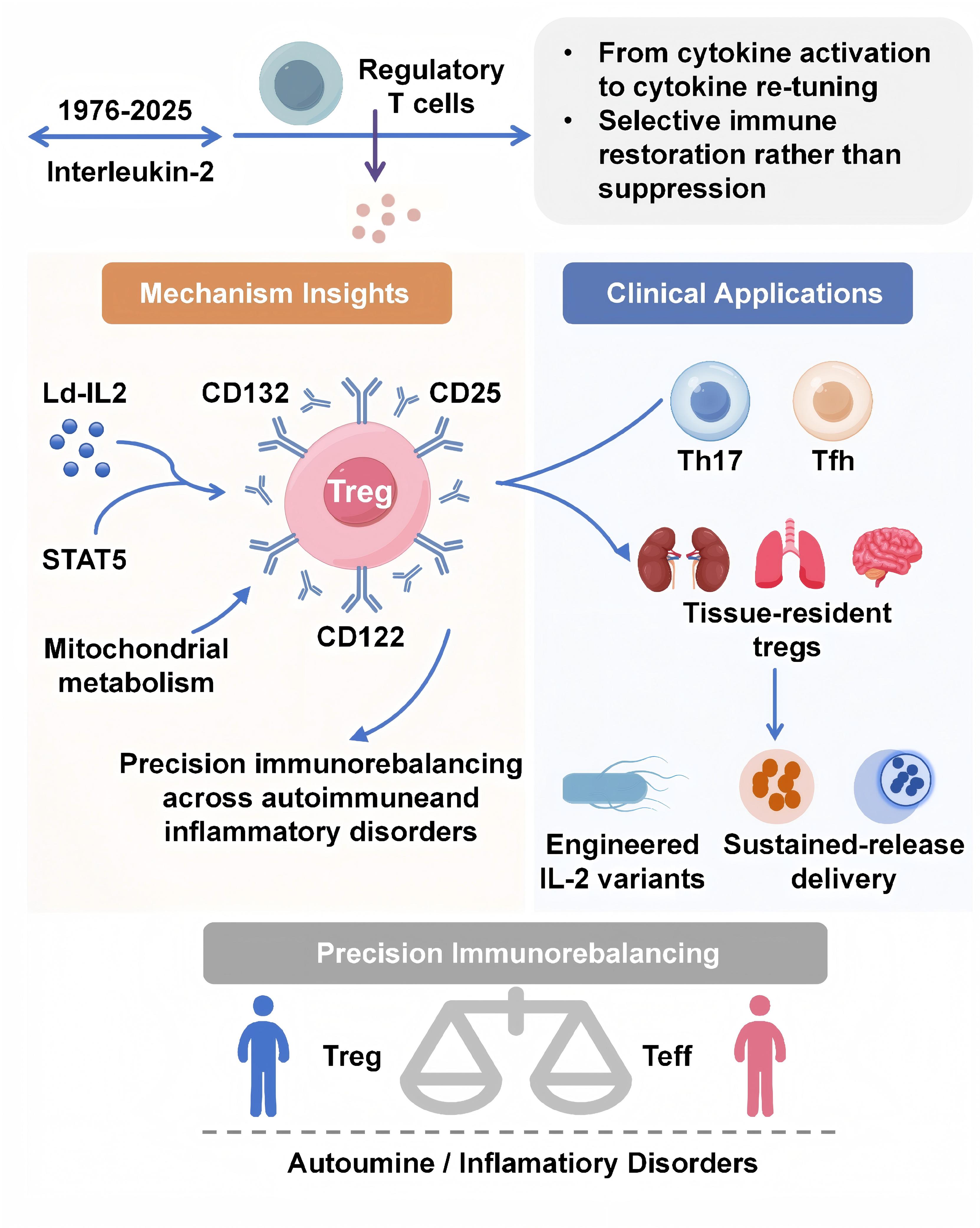
Regulatory T cells (Tregs) are the central guardians of immune tolerance, safeguarding against autoimmune and inflammatory damage through Foxp3-dependent transcriptional programs. Recent breakthroughs in precision immunotherapy have revived interest in low-dose interleukin-2 (Ld-IL-2), a cytokine-based strategy that selectively expands and activates Tregs via the high-affinity IL-2 receptor (CD25). This review summarizes emerging mechanistic insights into how Ld-IL-2 orchestrates multilevel immune rebalancing and highlights its translational progress from molecular engineering to clinical applications across autoimmune diseases. We integrated recent findings from cellular, metabolic, and systems immunology studies, together with our own multi-center clinical trial data, to outline the dynamic networks linking IL-2 signaling, Treg plasticity, and immune homeostasis. Ld-IL-2 exerts a dose-dependent biphasic effect on the immune system, selectively enhancing Treg survival and function while restraining pathogenic Th17, Tfh, and Teff subsets. Beyond classical STAT5-FOXP3 activation, recent studies reveal that IL-2 reprograms Treg metabolism toward oxidative phosphorylation, stabilizes Foxp3 epigenetic landscapes, and coordinates intercellular communication through exosomal and tissue-resident networks. Innovations in topologically engineered IL-2 variants and sustained-release delivery systems (e.g., polylactic-acid microsphere–exosome composites) further extend the precision and durability of Treg-directed therapy. Clinical evidence from SLE, Sjögren's disease, and relapsing polychondritis confirms robust immune restoration and favorable safety profiles within defined dose windows. By selectively activating the Treg axis and reprogramming immune homeostasis, low-dose IL-2 represents a paradigm for precision immunotherapy. Integrating molecular engineering and targeted delivery strategies will enable next-generation cytokine therapies to achieve durable immune tolerance across autoimmune and inflammatory diseases.
Keywords: Regulatory T cells; Low-dose Interleukin-2; Foxp3; Immune tolerance; Precision immunotherapy; Translational medicine
Introduction
Interleukin-2 (IL-2) was first identified in 1976 as a potent T-cell growth factor that sustains clonal expansion of activated lymphocytes and fuels effector immunity [1]. During the early decades of cytokine research, IL-2 was considered primarily as a molecule that amplifies cytotoxic T-cell and natural killer (NK) cell activity, leading to its clinical application in high-dose regimens for metastatic cancer and chronic viral infection [2]. However, the subsequent identification of a distinct CD4+T cell subset expressing high levels of the IL-2 receptor α-chain (CD25) and the transcription factor Forkhead box protein 3 (Foxp3) challenged this view. These cells—now known as regulatory T cells (Tregs)—were shown to depend critically on IL-2 for their development, survival, and suppressive function [3]. This paradigm shift transformed IL-2 from a mere activator of immune responses into a master regulator of immune tolerance.
In the decades since Shimon Sakaguchi and colleagues defined Tregs as essential for preventing spontaneous autoimmunity, IL-2 has emerged as the key cytokine linking effector and regulatory immunity [4]. Whereas high-dose IL-2 promotes effector proliferation through intermediate-affinity βγ (CD122/CD132) receptor signaling, low-dose IL-2 (Ld-IL-2) preferentially engages the high-affinity αβγ (CD25/CD122/CD132) receptor complex that is constitutively expressed on Tregs [5-6]. This receptor hierarchy provides a unique therapeutic window in which Ld-IL-2 selectively expands and stabilizes Tregs without activating pathogenic effector T cells or inducing generalized inflammation. Previous experimental and clinical studies have demonstrated robust Treg reconstitution and immune restoration across autoimmune conditions such as Systemic lupus erythematosus (SLE), primary Sjögren's disease (SjD), type 1 diabetes, and graft-versus-host disease [7-10].
Despite great advances in molecular immunology, treatment of autoimmune diseases still largely relies on broad immunosuppressive agents—glucocorticoids, calcineurin inhibitors, or cytotoxic drugs—that indiscriminately suppress both protective and pathogenic immunity. Although these regimens can alleviate symptoms, their long-term administration is associated with infection risk, metabolic toxicity, and incomplete disease remission. More importantly, they fail to address the underlying immune dysregulation characterized by the numerical and functional decline of Tregs and the overactivation of effector T cells. Thus, the unmet clinical requirement lies in restoring the physiological immune balance rather than merely dampening immune activation. Targeted Treg restoration through cytokine modulation represents a rational and biologically precise strategy to achieve this goal.
Consequently, the therapeutic strategy of "precision immunorebalancing" has been proposed. This approach aims to restore immune homeostasis by selectively modulating the immune system at cellular, molecular, and tissue levels. Ld-IL2 preferentially activates high-affinity receptors on Tregs to induce tolerance without global immunosuppression. Numerous researches revealed that its efficacy stems not only from Treg expansion but also from a fundamental reprogramming of Treg metabolism, epigenome, and transcriptome, which reinforces their suppressor identity and simultaneously attenuates Th17 and Tfh pathways. Additional modulation of NK and memory CD8⁺ T cells further contributed to immune homeostasis. Translationally, engineered IL-2 variants with extended half-life and controlled receptor selectivity, along with sustained-release delivery systems such as polylactic-acid microspheres or exosome-based nanocarriers, are further enhancing the precision and durability of Treg-targeted therapy.
In this review, we provide a comprehensive overview of the mechanistic underpinnings of Treg-centered IL-2 signaling and summarize recent translational advances that position Ld-IL-2 as a cornerstone of next-generation precision immunotherapy. By integrating molecular biology, systems immunology, and clinical evidence, we aim to delineate how IL-2 has evolved from a classic immunostimulant to a fine-tuned regulator of immune tolerance, opening new avenues for durable disease remission across autoimmune and inflammatory disorders.
Molecular Mechanisms of IL-2-Mediated Treg Regulation
IL-2 Receptor Signaling and the STAT5–FOXP3 Axis
The biological effects of IL-2 are determined by the composition and affinity of its trimeric receptor complex (Figure 1). Tregs constitutively express the high-affinity IL-2 receptor, composed of α (CD25), β
(CD122), and γ (CD132) chains, enabling them to respond to picomolar concentrations of the cytokine [5]. In contrast, conventional CD4+ and CD8+ T cells express only the intermediate-affinity βγ
complex, which requires much higher IL-2 levels for activation [11-12]. This
receptor hierarchy forms the molecular foundation for the selective responsiveness of Tregs to low-dose IL-2
(Ld-IL-2).
Figure 1. Mechanistic network of low-dose IL-2 in immune regulation. Low-dose IL-2 preferentially signals through the high-affinity IL-2 receptor (IL-2Rαβγ) on regulatory T cells, engaging JAK–STAT and mTOR–AMPK signaling. These pathways sustain Foxp3 transcription and reprogram cellular metabolism, thereby stabilizing Treg identity and function. The resulting network suppresses Th17 and Tfh responses while maintaining CD8+ T and NK cell homeostasis, collectively restoring immune balance.
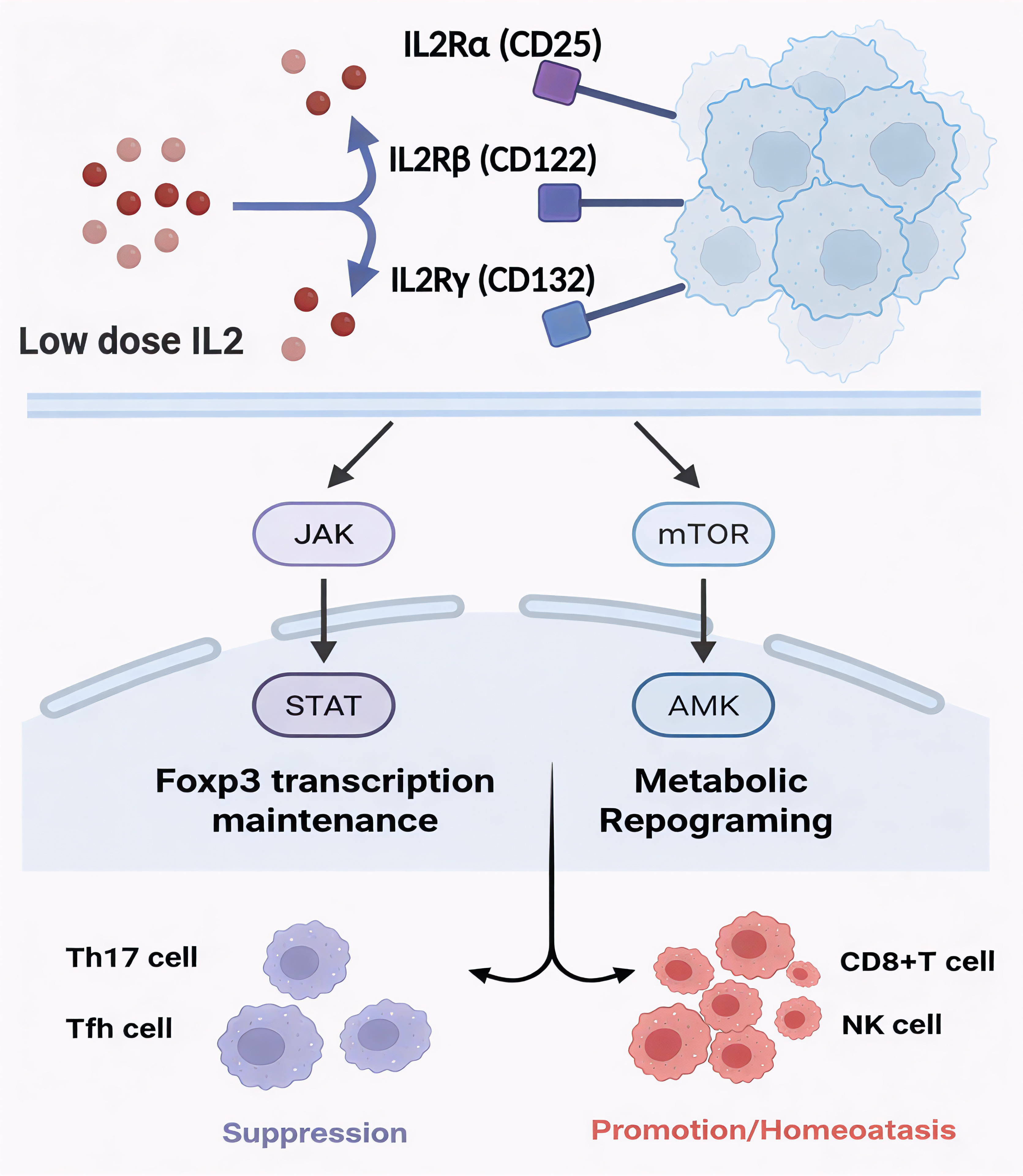
Upon IL-2 binding, JAK1 and JAK3 associated with CD122 and CD132 are activated, leading to phosphorylation of STAT5 [13-16]. Dimerized STAT5 translocates into the nucleus to bind FOXP3 and its enhancer regions, thereby reinforcing the transcriptional program that sustains Treg identity [17]. Concomitantly, PI3K–AKT and MAPK pathways undergo transient activation, delivering metabolic and pro-survival signals without converting Tregs into effector cells [18]. In contrast, excessive AKT activation—as induced by high-dose IL-2—can destabilize FOXP3 expression, highlighting the critical role of a controlled signaling amplitude [16].
FOXP3 acts as the lineage-defining transcription factor that suppresses pro-inflammatory genes while promoting immunosuppressive mediators such as IL-10, TGF-β, and CTLA-4 [19-20]. Phosphorylated STAT5 not only initiates FOXP3 transcription but also maintains its epigenetic accessibility through recruitment of histone acetyltransferases [21]. Recent multi-omics analyses show that sustained, low-intensity STAT5 signaling preserves FOXP3 stability, whereas intermittent or excessive activation promotes Treg plasticity and loss of function [22]. Thus, Ld-IL-2 achieves an optimal signal-strength threshold that sustains suppressive potency while avoiding effector conversion.
Moreover, IL-2 signaling influences the composition of the Treg pool. Peripheral IL-2 availability regulates the balance between thymus-derived Tregs and peripherally induced Tregs (pTregs), both essential for maintaining tolerance [23]. Through up-regulation of B cell lymphoma 2 (Bcl-2) and down-regulation of pro-apoptotic Bim, IL-2 prolongs Treg survival and enhances their tissue persistence [24-25]. Collectively, these findings underscore that IL-2 is not merely a growth factor but the master regulator orchestrating Treg lineage commitment, stability, and homeostatic renewal.
Epigenetic and Metabolic Reprogramming of Tregs
In addition to the canonical receptor signaling, IL-2 emerges as a critical regulator of the epigenetic and metabolic
programs that determine Treg identity. At the chromatin level, Ld-IL-2 maintains demethylation of the Treg-specific
demethylated region (TSDR) within the FOXP3 locus, which is the hallmark of stable, suppressive Tregs [26-29]. This demethylated configuration is stabilized by STAT5, which promotes
the recruitment of TET demethylases and suppresses DNA methyltransferase (DNMT) activity, preventing the loss of FOXP3
expression under inflammatory conditions [30-32]. Single-cell ATAC-seq studies
further reveal that IL-2 stimulation increases chromatin accessibility at genomic loci encoding critical Treg
functional molecules, including IL2RA, CTLA4, and IKZF2 (Helios), reinforcing the transcriptional network sustaining
Treg lineage fidelity [18, 33-35].
Metabolically, IL-2 signaling coordinates the balance between oxidative phosphorylation (OXPHOS) and glycolysis, a pivotal determinant of Treg function. In contrast to effector T cells that rely heavily on aerobic glycolysis, Tregs prefer fatty acid oxidation and mitochondrial respiration to support long-term survival and suppressive capacity [36-37]. Through controlled activation of the PI3K–AKT–mTOR axis, Ld-IL-2 enhances fatty-acid oxidation while limiting anabolic glycolysis [38]. Simultaneously, IL-2 up-regulates AMP-activated protein kinase (AMPK), promoting mitochondrial biogenesis and reactive-oxygen-species (ROS) detoxification [39-40]. Together, this IL-2-driven metabolic reprogramming enables Tregs to adapt to nutrient-poor or hypoxic microenvironments, which are frequently encountered in chronically inflamed tissues.
IL-2 signaling integrates with cellular metabolism to regulate the availability of epigenetic substrates, notably within one-carbon metabolism, which generates the universal methyl donor S-adenosylmethionine (SAM) [41-42]. This crosstalk ensures a sufficient supply of methyl-donor for sustaining FOXP3 chromatin marks [43]. Perturbation of this loop, for instance through mTOR hyperactivation or glucose overload, results in FOXP3 instability and Treg dysfunction [18, 44]. These insights explain why Ld-IL-2, but not higher doses, consistently promotes the expansion of functionally stable and long-lived Tregs.
Recent proteomic studies further indicate that IL-2 induces a shift toward an anti-inflammatory secretome, including soluble CD25 and adenosine-generating enzymes (CD39/CD73) [45]. Such extracellular metabolites contribute to a tolerogenic microenvironment that suppresses the activation of bystander effector T cells and antigen-presenting cell activation. Hence, IL-2 not only controls the intracellular transcriptional and metabolic programs of Tregs but also directs the remodeling of the extracellular immunological landscape, establishing a multilayered network of immune regulation.
Tissue Residency and Exosomal Communication
Emerging evidence indicates that IL-2 signaling extends beyond systemic immune regulation to spatially confined tissue
microenvironments [46]. Tissue-resident Tregs (TR-Tregs) in non-lymphoid organs including salivary
gland, lung, skin, and skeletal muscle exhibit distinct transcriptomic signatures characterized by high IL2RA and
IL1RL1 (ST2) expression. These TR-Tregs not only suppress inflammation but also participate in tissue repair through
secretion of amphiregulin and growth factors [47-50]. The density and functional
activity of TR-Tregs are critically dependent on local IL-2 availability, which can be derived from tissue dendritic
cells, natural killer (NK) cells, or exogenous low-dose IL-2 therapy. In murine models of Sjögren's-like exocrinopathy
and arthritis, targeted delivery of IL-2 to glandular or joint tissues enhances Treg accumulation, reduces Th17
infiltration, and restores tissue integrity [51-53], highlighting the
therapeutic potential of spatially restricted cytokine modulation.
A novel mechanism of IL-2-mediated communication involves extracellular vesicles (EVs) and exosomes. Activated T cells and stromal cells can package IL-2 and IL-2 receptor subunits into exosomes, forming a localized cytokine delivery system that extends the half-life and range of IL-2 signaling. Furthermore, Treg-derived exosomes carry immunoregulatory molecules such as CTLA-4, LAG-3, and miRNAs (miR-155, miR-21) that further suppress effector responses [54-56]. Recent studies suggest that Ld-IL-2 treatment enhances the production of these Treg exosomes, which can be detected in peripheral circulation and correlate with clinical remission in autoimmune diseases [57-60]. Such findings open a new dimension of intercellular communication where IL-2 not only acts as a soluble cytokine but also as a cargo within vesicular networks shaping the immune micro-environment.
These insights into tissue-specific IL-2 biology are informing the development of next-generation therapeutic formulations. Engineered delivery systems such as biomaterial-based slow-release microspheres and exosome-hybrid nanocarriers can deliver IL-2 directly to inflamed sites, maintaining effective local concentrations while minimizing systemic exposure [61]. This approach recapitulates the natural activity of IL-2 within tissue niches and enhances the functional persistence of resident Treg populations. Together, these mechanistic insights demonstrate that Ld-IL-2 acts not as a simple proliferative molecule but as a multidimensional signal integrating receptor affinity, epigenetic stability, metabolic fitness, and tissue-specific microenvironmental context to achieve precise immune rebalancing.
Systemic and Cellular Effects Beyond Tregs
The therapeutic efficacy of Ld-IL2 is initiated by its high-affinity binding to Tregs but culminates in the systemic recalibration of immunity. This stems from a coordinated reprogramming of the immune landscape, inhibiting effector T-cell and B-cell responses, while simultaneously regulating the homeostasis of innate and cytotoxic lymphocytes. The following discussion details how Ld-IL-2 integrates these multi-cellular processes to re-establish systemic immune tolerance (Table 1).
The Treg–Th17/Tfh Counter-regulatory Axis
The pathogenesis of autoimmune inflammation frequently involves a dysregulated balance between immunosuppressive
regulatory T (Treg) cells and pathogenic T helper 17 (Th17) and T follicular helper (Tfh) cells. Th17 cells produce
IL-17A, IL-21 and GM-CSF, driving tissue infiltration and neutrophil recruitment, whereas Tfh cells promote B-cell
activation and autoantibody production [62-64]. The reciprocal relationship
among these subsets is strongly regulated by interleukin-2 (IL-2) signaling.
At the molecular level, IL-2 serves as a potent negative regulator of Th17 and Tfh differentiation. Low dose of IL-2 selectively activates STAT5, which competitively antagonizes STAT3 at key genomic loci, including the promoters of IL17A and the Tfh master regulator BCL6, thereby suppressing the transcriptional programs for inflammatory cytokines and Tfh lineage genes [65-67]. Consequently, Ld-IL-2 not only promotes Treg stability but also actively constrains the differentiation of Th17 and Tfh lineages. This dual mechanism yields a synergistic anti-inflammatory effect: Treg-derived IL-10 and TGF-β suppress residual Th17 activity, while the concomitant reduction in Tfh-derived IL-21 reduces B-cell stimulation and autoantibody generation [68-69].
Clinical and pre-clinical evidence consistently support this mechanistic framework. In patients with systemic lupus erythematosus (SLE) and Sjögren's disease (SjD), Ld-IL-2 therapy consistently increases circulating Treg numbers while concomitantly reducing the frequencies of Th17 and T follicular helper (Tfh) cells, resulting in improvements in disease activity [8-9, 70-71]. Corresponding observations in various mouse models demonstrate that these changes correlate with suppressed germinal center reactions and decreased autoantibody titers [72-73]. Thus, by modulating the Treg–Th17/Tfh axis, Ld-IL-2 effectively redirects the adaptive immune response from a pathogenic inflammatory state toward controlled tolerance.
The suppression of Th17/Tfh pathways exhibits a clear dose-dependency. At an optimal low dose (approximately 1 million IU), IL-2 robustly expands the Treg compartment without significantly activating effector T cells (Teffs) [7, 60, 74]. In contrast, higher doses (> 3 M IU) can lead to the partial activation of effector pathways, reflecting the biphasic response to IL-2 that has been documented in clinical dose-related studies [10]. Therefore, precise dose decision is essential to retain the desired balance between regulatory and effector immunity.
Table 1. Differential effects of IL-2 on immune cell subsets and functional outcomes.
| Cell Subset | IL-2 Receptor Expression | Response to Low-dose IL-2 | Functional Effect | Impact on Immune Homeostasis |
|---|---|---|---|---|
| Treg (CD4+CD25+FOXP3+) | CD25 high CD122+ CD132+ | Strong activation (↑ STAT5) | ↑ Suppressive cytokines (IL-10, TGF-β) | Restores tolerance |
| Teff (CD4+ effector) | CD25 low | Minimal effect | ↓ IFN-γ production | Reduces inflammation |
| Th17 | CD25 low | Indirect inhibition via Treg | ↓ IL-17 output | Prevents autoimmunity |
| Tfh | CD25 low | Suppressed | ↓ B cell help | Decreases autoantibody |
| CD8+ memory-like T | CD122 high | Moderate activation | ↑ Cytotoxic homeostasis | Enhances infection control |
| NK cells | CD122 high | ↑ Survival and cytotoxicity | Balanced innate response | Maintains immune surveillance |
CD8+ T-cell Regulation and Memory-like Differentiation
Beyond CD4+ T cells, IL-2 also plays a crucial role in the cytotoxic compartment. Ld-IL-2 preferentially expands a
distinct population of CD8+CD122+ memory-like T cells that express intermediate-affinity IL-2 receptors (CD25) [75-76]. This subset is transcriptionally defined by the concurrent upregulation
of Eomesodermin (Eomes) and the anti-apoptotic protein Bcl-2, a molecular signature that supports their long-term
persistence and confers a regulated cytotoxic potential, without inducing excessive tissue damage [77-79].
Mechanistically, Ld-IL-2 engages the βγ receptor complex (CD122/CD132) on these cells to promote homeostatic proliferation and IL-10 production, contributing to the resolution of inflammation [80-82]. Furthermore, IL-2 modulates the memory differentiation pathway of CD8+ cells. Continuous low-level signaling via STAT5 favors a central-memory phenotype (CD62L+CCR7+) over short-lived effector cells [83-84]. This phenotype ensures a rapid but non-pathogenic response upon secondary antigen exposure, which is especially relevant for patients with chronic viral infections or immune deficiency associated with autoimmunity. Collectively, these findings illustrate how Ld-IL-2 fine-tunes the cytotoxic arm of immunity to support immune equilibrium rather than immunosuppression.
Modulation of NK Cell Activity and Innate Immunity
Natural killer (NK) cells represent a pivotal innate immune target of IL-2. Constitutively expressing the
intermediate-affinity IL-2 receptor βγ complex (CD122/CD132), NK cells respond to both high- and low-dose IL-2, albeit
with divergent functional consequences [85-86]. At low doses, IL-2
preferentially enhances the CD56bright regulatory NK subset that produces IL-10 and modulates antigen-presenting cells
[85, 87-90]. These cells exhibit reduced cytotoxic activity
but increased immunoregulatory potential, providing an additional layer of immune control.
Experimental studies show that Ld-IL2 upregulates the expression of inhibitory receptors such as NKG2A and CD94 on NK cells, thereby raising the threshold for activation and preventing excessive cytolytic activity, while preserving critical antiviral functions like interferon-gamma (IFN-γ) secretion [91-92]. This "calibrated activation" state supports tissue repair and homeostasis as high-dose IL-2 triggers the expansion of CD56dim cytolytic NK cells, which can exacerbate tissue damage and cytokine toxicity [93]. Thus, the NK cell response mirrors the dose-dependent duality of IL-2 seen in T cells.
Furthermore, IL-2 indirectly modulates innate immunity through Treg-educated mechanisms. Tregs expanded by Ld-IL-2 secrete copious amounts of IL-10 and TGF-β, which skew macrophage polarization toward an M2-like, tissue-reparative phenotype and restrain the pro-inflammatory capacity of dendritic cells [94-95]. This cascade establishes a positive feedback loop in which innate cells further support Treg maintenance and local IL-2 availability, stabilizing the immunological microenvironment.
Such innate re-education may explain the clinically favorable infection profile associated with Ld-IL-2 therapy. Unlike broad-spectrum immunosuppressants which increase susceptibility to opportunistic infections, Ld-IL-2 has not been linked to elevated infection rates in clinical trials [73, 96-98]. It enhances mucosal barrier immunity and NK-mediated viral clearance. These observations underscore that precision cytokine therapy can restore tolerance without compromising host defense.
Integrated Network of Immune Homeostasis
Ld-IL2 develops a coordinated recalibration of the immune system by engaging regulatory T cells (Tregs), effector T
cells, cytotoxic lymphocytes, and innate subsets within an integrated network of homeostatic control [99]. Rather than acting as a generalized immunosuppressant, Ld-IL-2 restores a dynamic equilibrium
that preserves immune surveillance while restraining autoimmunity. This fine-tuned network operates through multiple
interdependent layers.
At the signaling level, regulatory T cells (Tregs) express high-affinity IL-2Rαβγ complexes, which allow them to efficiently respond to low concentrations of IL-2 and establish a hierarchical access to this cytokine [4, 16, 100]. Through this preferential capture of IL-2, Tregs act as a critical sink that shapes the cytokine landscape and modulates effector cell activation thresholds. In parallel, feedback mechanisms reinforce this dominance as Tregs secrete IL-10 and generate adenosine through the CD39/CD73 axis, suppressing effector and antigen-presenting cell (APC) activation while limiting IL-2 consumption by competing lymphocyte subsets [101]. These negative feedback loops sustain the regulatory pool and prevent exhaustion of the cytokine niche. Beyond local regulation, cross-compartmental communication connects systemic and tissue-localized tolerance programs [102]. Tregs communicate with innate immune cells via cytokines, exosomes, and metabolites, thereby synchronizing peripheral and organ-specific tolerance.
Through these integrated mechanisms, low-dose IL-2 acts as a biological "immune thermostat," recalibrating the immune set-point toward tolerance rather than non-selective suppression, in contrast to conventional immunosuppressive strategies that broadly inhibit immune activity [12].
Conceptual Implications
The systemic immunomodulatory effects of Ld-IL-2 suggest that regulatory immunity is not confined to a single cell
type but arises from a dynamically integrated network of interdependent signals. Ld-IL-2 operates by coordinately
stabilizing the regulatory T cell (Treg) compartment while concurrently reprogramming the function of effector T cells
and innate immune populations [4, 12]. This understanding has shifted the
therapeutic paradigm from cytokine replacement to cytokine re-tuning, using low and precisely calibrated doses to
activate selective pathways that restore systemic balance [103].
The consistent clinical benefits observed with Ld-IL-2 across a spectrum of autoimmune diseases, from systemic lupus erythematosus to alopecia areata, provide compelling translational validation for this network-centric view of immune homeostasis [104-108]. Consequently, Ld-IL-2 is rationally viewed as the foundational therapeutic agent in the emerging discipline of precision immunorebalancing, a field dedicated to the targeted restoration of immune homeostasis rather than its broad suppression.
Clinical Translation of Low-dose IL-2 Therapy
Dose-dependent biphasic responses and therapeutic window
The translational development of Ld-IL2 is rooted in its biphasic dose-response immunobiology. Pioneering oncology
studies using high-dose IL2 (> 10 MIU/day) demonstrated potent anti-tumor immunity but were hampered by severe
toxicity, including vascular leak syndrome, due to excessive activation of effector T and NK cells. In contrast,
experimental immunology revealed that picomolar concentrations (0.3–3 MIU) selectively expand Tregs without activating
pro-inflammatory Teff activation or systemic inflammation (Figure 2) [10]. This
bell-shaped relationship defines a narrow therapeutic window that is central to precision cytokine therapy [103, 109].
Figure 2. Dose-dependent biphasic effects of IL-2 on Treg cells. Low-dose IL-2 induces a selective, dose-dependent expansion of CD4+CD25hiFoxp3+ Tregs through engagement of the high-affinity IL-2Rαβγ complex, with maximal response at approximately 1×106 IU. Higher doses may activate conventional T (Tconv) and NK cells, thereby diminishing Treg selectivity and shifting the immune balance toward activation.
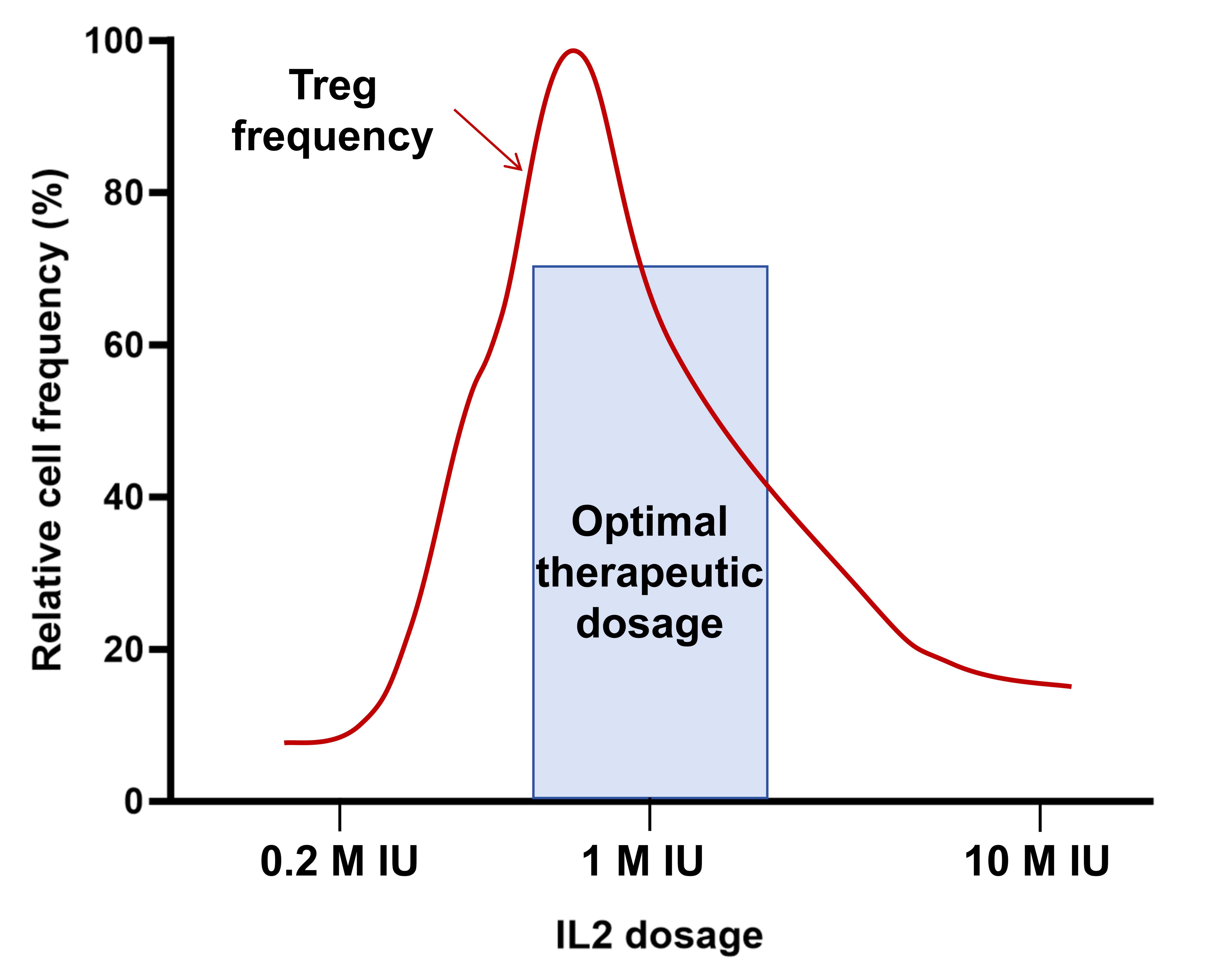
This selective effect is mechanistically controlled by differential sensitivity to IL-2. Tregs, owing to their constitutive expression of the high-affinity IL-2 receptor α-chain (CD25) and a primed JAK-STAT signaling apparatus, activate STAT5 at concentrations 100- to 300-fold lower than those required by Teffs [110-111]. Clinically, trials comparing 1 MIU versus 3 MIU dosing confirm this principle, demonstrating that a dose of 1 MIU optimally expands Tregs and yields clinical improvement with minimal adverse events, whereas a 3 MIU dose can induce transient Teff activation and associated side effects like fatigue [8, 10, 105, 112]. Importantly, pharmacodynamic analyses further demonstrate that serum IL-2 concentrations return to baseline within hours after Ld-IL-2 administration, allowing physiologic cycling of Treg signaling without receptor desensitization.
Longitudinal data reveal that the degree of Treg expansion is a strong predictor of clinical efficacy, with patients achieving a doubling (≥200%) in Treg frequency typically exhibiting sustained disease remission, supporting Treg frequency as a pharmacodynamic biomarker for dose optimization [60, 71, 113]. These quantitative data provide a foundation for precision dosing models integrating IL-2 pharmacokinetics, baseline Treg status, and cytokine network feedback.
Clinical evidence across autoimmune diseases
Systemic Lupus Erythematosus (SLE)
SLE represents the prototype disease in which Ld-IL2 therapy has
demonstrated consistent efficacy. Multiple open-label and randomized trials have reported significant improvements in
SLEDAI scores accompanied by marked Treg expansion. In our multicenter double-blind phase IIb trial (NCT04077684),
patients receiving 1 MIU IL-2 for 5 consecutive days per cycle showed a 2.2-fold increase in Treg frequency and a mean
4-point reduction in SLEDAI after 12 weeks [8, 60, 108, 114-115]. Transcriptomic profiling revealed down-regulation of type-I
interferon-responsive genes and normalization of IL-21 and CXCL13 signatures, reflecting suppression of Tfh-driven
autoantibody production [72, 116]. The favorable safety profile was confirmed,
with only mild adverse events (fatigue, transient erythema < 5%) and no increased infection risk.
Primary Sjögren's Disease (SjD)
In Sjögren's disease, Ld-IL-2 has shown promise as a disease-modifying
therapy capable of restoring immune balance and exocrine gland function. A prospective study in 30 patients
demonstrated that treatment significantly increased the Treg/Th17 ratio and improved unstimulated salivary flow
rates following two cycles [9]. Flow cytometric analysis showed preferential expansion of
CCR4+CXCR5- Tregs and reduction of circulating Tfh cells. These immunological changes were associated with
clinical benefit, as evidenced by a >3-point reduction in ESSDAI scores in 70% of participants.
Relapsing Polychondritis (RP)
Relapsing polychondritis (RP), a rare autoimmune disorder characterized
by steroid-dependent cartilage inflammation, presents a compelling target for Ld-IL-2 therapy [117]. In our exploratory open-label trial (n = 9), 1 MIU IL-2 for 5 days per cycle led to rapid
symptom improvement within two weeks, with normalization of C-reactive protein (CRP) levels and Relapsing
Polychondritis Disease Activity Index (RPDAI) scores in the majority of patients.
Type 1 Diabetes and Other Conditions
The application of Ld-IL-2 extends to other autoimmune contexts.
In new-onset type 1 diabetes, therapy aims to preserve residual β-cell function, with studies showing that Ld-IL-2
can increase Treg frequency and stabilize C-peptide levels [10, 118].
Promising pilot data in autoimmune vasculitis and hepatitis further underscore its potential as a broad-spectrum
immunomodulator [119-120]. Collectively, across this spectrum of diseases,
Ld-IL-2 consistently achieves a quantifiable restoration of Treg populations (typically a 150–250% increase) and
delivers clinically meaningful disease amelioration with a minimal toxicity profile.
Safety and tolerability profile
The collective safety data from over 600 patients treated with Ld-IL-2 worldwide reveal an exceptionally favorable
profile (Table 2). The most common adverse events are mild and transient, including
injection-site reactions, fatigue, and mild nausea, with an incidence generally below 10%. Critically, no grade ≥2
cytokine-release syndrome or treatment-related serious infections have been consistently reported, and routine
laboratory parameters (e.g., hepatic and renal function) remain stable. Immune monitoring confirms that Ld-IL-2
does not compromise protective immunity, as evidenced by preserved antiviral NK-cell function and vaccine
responses [121]. These results contrast with those of conventional immunosuppressants and
underscore the fundamental distinction between precise immune recalibration and generalized immunosuppression.
Innovative formulations and delivery strategies
Topologically engineered IL-2 variants
Advances in protein engineering have yielded a newly developed IL-2 variant designed to achieve superior cellular
selectivity[122-124]. These molecules are engineered by our group to
retain high affinity for the CD25 (IL-2Rα) subunit while attenuating binding to CD122 (IL-2Rβ), thereby favoring
signaling through the high-affinity receptor complex predominantly expressed on Tregs. This design not only
enhances Treg selectivity but also often extends serum half-life, mitigating the need for frequent dosing.
Preclinical studies with such variants have demonstrated sustained Treg expansion for over five days, positioning
them as promising second-generation candidates for long-acting immune recalibration.
Sustained-release biomaterial platforms
To address the inherently short plasma half-life of native IL-2, significant efforts have been directed toward
developing sustained-release delivery systems. Biodegradable polylactic-acid (PLA) microspheres can encapsulate
IL-2 and provide controlled release of the bioactive cytokine over 72 to 96 hours, maintaining stable, physiologic
concentrations and avoiding the peaks associated with toxicity[125]. Further sophistication is
achieved by combining these microspheres with Treg-targeting exosomes to create hybrid complexes, which can
facilitate the targeted delivery of IL-2 to inflamed tissues such as salivary glands or joints. This approach
achieves localized Treg reconstitution and tissue repair with negligible systemic exposure. Pre-clinical data
demonstrate marked reduction of inflammatory cell infiltration and enhanced tissue integrity in models of
Sjögren-like syndrome and arthritis.
Localized delivery and minimally invasive routes
Given the emerging importance of tissue-resident Tregs, local subcutaneous or periglandular administration is
being explored. Preliminary clinical studies from our group indicate that parotid region micro-injection achieves
regional Treg accumulation and amelioration of glandular inflammation in SjD patients. Such approaches leverage
physiological IL-2 gradients within tissue niches and hold the potential to reduce systemic dose requirements.
Together, these formulation and delivery innovations represent the translational frontier of precision cytokine
therapy.
Table 2. Major clinical trials and observational studies of low-dose IL-2 in autoimmune diseases.
| Disease | Study Type / Year | Sample Size | Dose (million IU) / Schedule | Clinical Outcome | Safety Profile |
|---|---|---|---|---|---|
| SLE | Double-blind RCT [8] (NCT02465580 and NCT02932137) | 60 | 1×every other day for 2 weeks and followed by a 2-week break /cycle | Improved SRI-4 response rate | Lower incidence of infection |
| SLE | Multicenter Phase II RCT [108] (NCT02955615) | 100 | 1.5 daily × 5 days, then weekly | SLEDAI −5.1; reduced flares | Transient fatigue (5%) |
| SLE | Ongoing Phase II (Unpublished; NCT04077684) | 80 | 0.2–1 dose every other day × 12 weeks, then weekly for 12 weeks | Improved SRI-4 response | Well tolerated |
| pSS | Randomized trial [9] | 30 | 1×every other day for 2 weeks and followed by a 2-week break /cycle | ESSDAI −3 | No serious AEs and lower infection risk |
| RP | Pilot RCT (Unpublished; NCT04077736) | 10 | 1 × 5 days, then weekly | RPDAI −9; No AEs | |
| T1DM | DIL T1D trial (NCT01827735) [112] | 40 | 0.04×10⁶ to 1.5×10⁶ IU/m² daily | Increases in Treg frequencies | None reported |
| T1DM | Phase I/II RCT (DILfrequency, NCT01353833) [10] | 24 | 0.3–3 × 5 days, adaptive dosing | ↑ β-cell function; ↓ HbA1c | no serious AEs |
| GVHD | open-label (NCT01517347) [152] | 90 | 1.0 × 10⁶ IU/m² daily× 14 days | Minimal residual disease-positive | None reported |
| GVHD | observational cohort (NCT00529035) [7] | 29 | 0.3/1/3× 10⁶ IU/m² × 8 weeks | preferential, sustained Treg cell expansion | None reported |
| Alzheimer's disease | Phase 2a RCT (NCT06096090) [153] | 38 | 1.0 × 10⁶ IU/day×21 weeks | CSF Aβ42 levels ↑ | None reported |
| Amyotrophic lateral sclerosis | MIROCALS study (NCT03039673) [154] | 304 | 2 × 10⁶ IU/day× 5 days | Decrease in risk of death | No serious AEs |
| Bullous pemphigoid | Perspective study (ChiCTR2000028707) [97] | 43 | half million IU every other day × 8 weeks | Shorter disease control time | No serious infections |
Biomarkers and precision monitoring
The translational development of Ld-IL2 has been greatly facilitated by the evolution of quantitative
immunomonitoring assays. Flow cytometric measurement of CD25+CD127low FOXP3+ Tregs remains the gold-standard
biomarker, but emerging omics approaches providing unprecedented resolution [126-127]. Single-cell RNA and TCR sequencing have identified distinct Treg clonotypes expanding
after therapy, while epigenetic analyses of FOXP3 locus demethylation (e.g., TSDR methylation status) allow for
longitudinal tracking of Treg lineage stability [128-130]. Circulating
exosomal IL-2 and microRNA signatures (miR-155, miR-21) correlate with clinical response and may serve as
minimally invasive surrogate biomarkers [131]. The integration of these multidimensional data
into AI-driven models is poised to enable personalized dose optimization and prediction of long-term treatment
efficacy.
Comparative advantages over traditional immunosuppression
Ld-IL-2 possesses distinct mechanistic and clinical advantages over conventional broad-spectrum
immunosuppressants. Unlike these agents, which non-specifically inhibit lymphocyte activation or proliferation,
Ld-IL-2 acts by selectively regulating the immune system. This mechanistic precision underlies several distinct
clinical advantages as Ld-IL2 restored physiological tolerance through a normalized Treg-to-effector T cell
balance and preserved antimicrobial immunity stemming from maintained NK and memory CD8+ T cell activity. The
re-establishment of a stable immune set-point underpins the potential for achieving drug-free remission in certain
individuals. These distinctive properties collectively position Ld-IL-2 not as a mere immunosuppressant, but as a
pioneering agent for immune reconstruction.
Future perspectives in translational design
The future trajectory of Ld-IL-2 therapy lies in the development of integrated platforms that synergize engineered
cytokine variants, advanced biomaterial-based delivery systems, and digital immunomonitoring (Figure
3 and Table 3). Multi-omics profiling can define patient-specific immune signatures that
predict responsiveness to IL-2. Strategic combinations of Ld-IL-2 with other immunomodulators [75, 96, 132-134] including JAK
inhibitors, microbiome-derived metabolites, or belimumab may achieve synergistic efficacy by concurrently
targeting complementary pathways. Ultimately, the goal is to develop a personalized immunotherapy ecosystem where
cytokine dosing is dynamically optimized based on real-time immune feedback, thereby transforming Ld-IL-2 from a
disease-specific intervention into a universal platform for restoring immune homeostasis across autoimmunity,
chronic inflammation, and transplantation medicine.
Figure 3. Engineered IL-2 variants and targeted delivery strategies. (A) Topological Engineered IL2 Variants. Rational IL2 mutation to reduce IL-2Rβ affinity while preserving IL-2Rα binding, achieving CD25-biased agonists with enhanced Treg selectivity, prolonged half-life, and higher stability. (B) Targeted Delivery for Sustained Release. A hybrid nanoparticle platform combining PLA microspheres and exosomes enables designed for controlled IL-2 release, maintaining therapeutic concentrations within the selective window. (C) Tissue-Selective Immunomodulation. Local injection including salivary, gland and joint concentrates IL-2 at sites of inflammation, maximizing Treg enrichment and resulting in reduced inflammation with limited systemic toxicity.
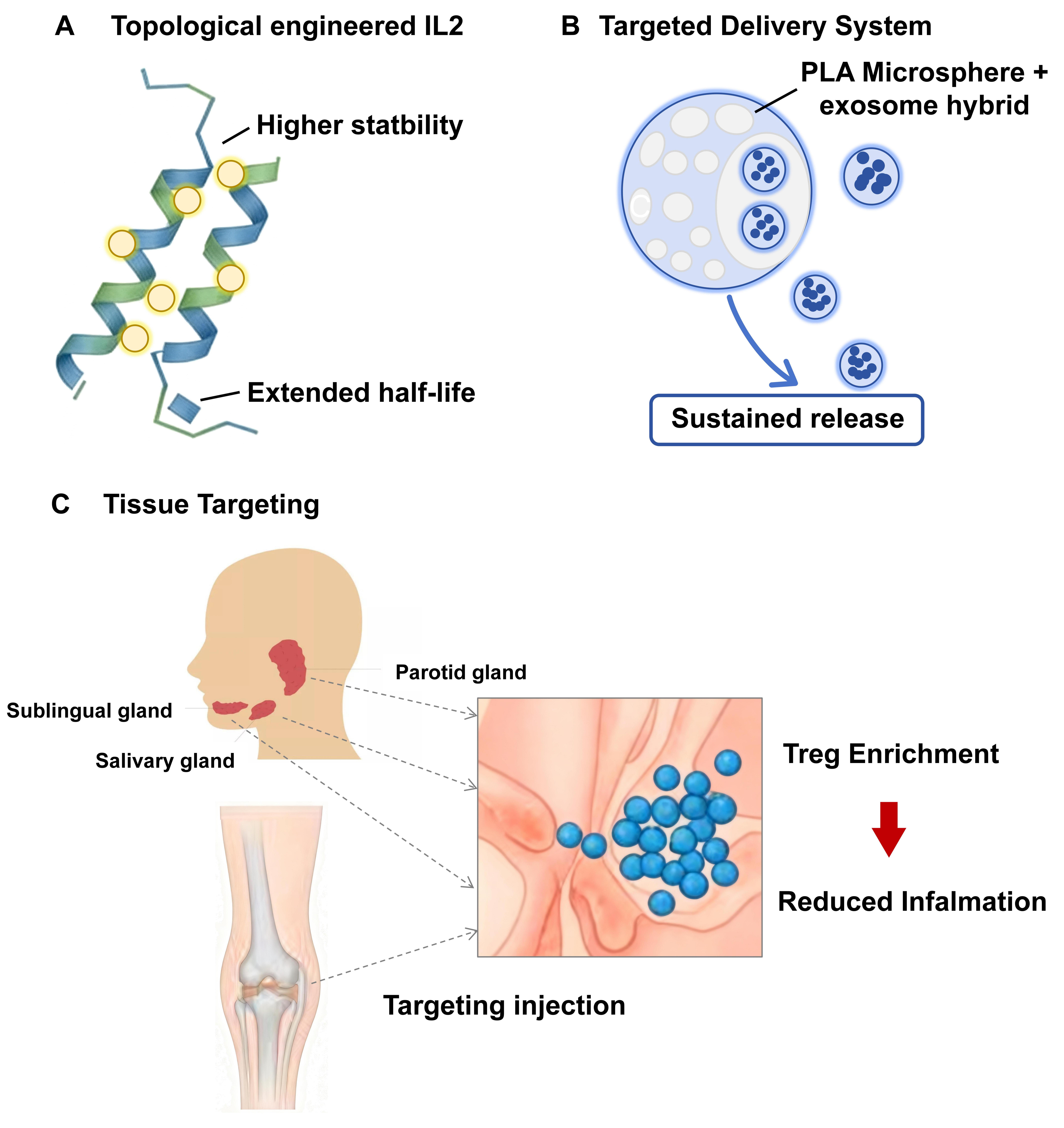
Table 3. Summary of engineered IL-2 variants and delivery systems.
| Approach | Design Feature | Mechanistic Advantage | Pre-clinical Outcome | Translational Potential |
|---|---|---|---|---|
| Topological engineered IL-2 | Reduced CD122 binding, retained CD25 affinity | Enhanced Treg selectivity | Prolonged half-life (> 48 h) | Phase I preparation |
| PEGylated IL-2 [122] | PEG chain modification | Reduced renal clearance | Improved AUC × 3 | Safety proven |
| IL2-4M-PEG [155] | Substitute amino acids and implement site-directed PEGylation | reduced CD25 binding activity and an extended half-life | Amplify effector cells | Enhance combined cancer therapies |
| IL-2/anti-IL-2 complex | Transient complex with anti-IL-2 mAb | Directed Treg bias | ↑ Treg/Teff ratio | Translatable |
| Pegylated rhIL-2 (SAR444336) [156] | Site-specifically pegylated form | enhanced PK and induced Tregs | reduced stimulation of off-target effector T and NK cells | Reduced inflammation in a delayed-type hypersensitivity model |
| PD1-IL2v [61] | Antibody-cytokine fusion proteins | Expansion of CD8+ and Tconv cells | ↑ cytotoxic CD8+ T cells | Next-generation immunocytokines |
| PLA microsphere system [157] | Slow-release (>72h) | Sustained serum level | Stable Treg expansion | Ready for pilot test |
| IL-2 mutein [158] | Fc-fused homodimer | reduced potency and enhanced Treg cell selectivity | Preferential Treg cell enrichment | Strong future potential |
| Exosome nanocarrier | Natural membrane vector | Targeted tissue delivery | Localized immunomodulation | Strong future potential |
Treg modification and clinical practice
Maintaining long-term Treg stability
The durability of Treg-mediated immune tolerance depends on the sustained expression and epigenetic stability of
FOXP3. Under inflammatory or metabolic stress, Tregs can undergo functional reprogramming—often termed "Treg
plasticity"—characterized by loss of FOXP3 expression and acquisition of pro-inflammatory, effector-like
properties [135-136]. In chronic autoimmune diseases, pro-inflammatory
cytokines such as IL-6, IL-1β, and TNF-α contribute to this instability by disrupting STAT5 signaling and
modifying the chromatin landscape at the FOXP3 locus, thereby impairing its transcription [137]. Consequently, a subset of expanded Tregs may adopt pathogenic effector or Th17-like
phenotypes, ultimately compromising the durability of therapeutic responses.
To address this challenge, several strategies are under exploration. Optimized dosing regimens, such as periodic administration of Ld-IL-2, may better support Treg lineage stability than continuous exposure, as intermittent pulses mimic physiological cytokine exposure [8, 59, 70]. Combining Ld-IL-2 with therapies that reinforce FOXP3 stability—such as low-dose rapamycin, histone deacetylase (HDAC) inhibitors, or metabolic modulators targeting the AMPK-mTOR pathway—has shown synergistic effects in preclinical studies. Advances in synthetic biology also allow enforced FOXP3 expression or CRISPR-mediated editing of important enhancer regions to generate functionally stabilized Treg cells. These engineered cellular therapies, while technically demanding, hold promise for achieving durable immune tolerance that persists beyond transient cytokine stimulation.
Inter-individual variability and biomarker-guided precision dosing
Substantial inter-individual heterogeneity in response to standardized Ld-IL2 regimens has emerged as a key
challenge in the clinic. This variability is influenced by genetic and immunological factors, including
polymorphisms in genes such as IL2RA, STAT5B, and PTPN2, which can alter IL-2 receptor sensitivity and downstream
signaling fidelity [138-141]. Furthermore, baseline immunological
responses including the frequency and activation state of regulatory T cells (Tregs) vary considerably across
different autoimmune diseases, contributing to divergent therapeutic outcomes [7, 106, 109].
Addressing this variability requires the development of quantitative biomarkers for real-time dose adjustment. The Treg/Teff ratio and absolute Treg counts serve as practical, short-term pharmacodynamic markers. These include the analysis of FOXP3 Treg-specific demethylated region (TSDR) methylation status, measurement of serum IL-2 trough levels, and assessment of phospho-STAT5 induction dynamics in responsive cell populations. Integrating these measurements into algorithm-driven dosing models could enable personalized treatment cycles that maintain each patient within their optimal "immune tolerance window." Ultimately, artificial intelligence platforms that synthesize longitudinal immunophenotyping, cytokine pharmacokinetics, and clinical data are expected to make precision dosing a practical reality.
Optimization of cytokine delivery and tissue targeting
The therapeutic application of native interleukin-2 (IL-2) is constrained by its rapid clearance from plasma, with
a half-life of merely 5-10 minutes, and its non-specific tissue distribution. These properties limit its efficacy
in autoimmune conditions where pathology is confined to specific organs. A central objective in the field is,
therefore, to achieve sustained, localized cytokine exposure while minimizing systemic diffusion and associated
off-target effects. Innovative biomaterial-based delivery systems offer a promising strategy to overcome these
limitations. Platforms such as poly (lactic acid) (PLA) microspheres, hydrogel depots, and exosome-hybrid
nanocarriers are designed to provide controlled release of IL-2, thereby extending its in vivo bioavailability,
reducing the frequency of administration, and facilitating enhanced accumulation at target sites [142-143].
A more advanced approach involves the development of formulations engineered for local activity within particular organs [144]. Periglandular injection of microsphere-encapsulated IL-2 has been shown to enrich Tregs locally and restore salivary function in models of Sjögren's disease, while intra-articular delivery holds potential for managing rheumatoid arthritis by modulating the joint microenvironment [145-147]. Combining spatial targeting with molecular engineering like topological IL-2 variants exhibiting selective CD25 binding could maximize therapeutic precision and minimize systemic effects [124]. The successful clinical translation of these sophisticated bio-engineered biologics will be closely tied to the ongoing evolution of regulatory frameworks tailored to assess their unique profiles.
Balancing immune tolerance and host defense
An inherent challenge of all immunomodulatory strategies is preserving protective immunity while inducing
tolerance. Although clinical trials have not reported increased infection rates with Ld-IL-2, long-term
surveillance is warranted, particularly in immunocompromised patients. Maintaining the functional capacity of
natural killer (NK) cell and memory CD8+ T cell activity is crucial for robust antiviral and anti-tumor
surveillance [76, 77, 82, 86]. Future
therapeutic protocols could incorporate adaptive dosing strategies informed by real-time biomarkers of these
effector functions, such as NK cell interferon-gamma (IFN-γ) production or CD8+T cell granzyme B levels, to
dynamically ensure that host defense remains uncompromised.
In parallel, the interplay between Ld-IL-2 and other immunotherapeutic modalities, such as vaccination and cancer immunotherapy, presents a compelling area for future investigation [4]. Emerging evidence suggests that intermittent Ld-IL2 could enhance vaccine responses by preserving immune memory, while engineered IL-2 antagonists or dual-function molecules may allow temporal toggling between tolerance and activation depending on clinical context [124].
Integrating multi-omics and system immunology
The complexity of IL-2 signaling networks calls for comprehensive multi-omics approaches to fully delineate its
impact on the immune system. Single-cell transcriptomics, chromatin accessibility mapping, and proteomics have
already delineated the cellular heterogeneity of Treg and Teff subsets responding to Ld-IL-2 [148]. Integrating these datasets with metabolomic profiles and microbiome analysis will yield a
more holistic understanding of the cytokine-induced remodeling of immune homeostasis. Subsequently, systems
immunology frameworks can leverage this integrated information to identify predictive molecular modules associated
with clinical response and resistance, thereby informing the development of rational combination therapies and
novel biomarker discovery [149].
This systems-level insight paves the way for rationally designed combination strategies. As co-administration of Ld-IL-2 with microbiota-derived short-chain fatty acids, which are known to promote Treg generation and function through epigenetic and metabolic mechanisms, could synergistically enhance immune tolerance [150]. Similarly, combining Ld-IL-2 with agents that neutralize type I interferon signaling may suppress the inflammatory condition that undermines Treg stability in certain autoimmune diseases [151]. Such "rational combination immunotherapies" embody the principle of precision immunorebalancing, aiming to recalibrate entire pathological networks rather than isolated targets.
Future design of next-generation IL-2 therapeutics
The future of IL-2-based therapy is set to be defined by the convergence of advanced protein engineering,
computational design, and intelligent delivery. Next-generation IL-2 variants are being engineered with structural
modifications that precisely control receptor subunit bias—favoring CD25 for Treg selectivity—while simultaneously
improving pharmacokinetic profiles and enabling effective signaling at near-physiological concentrations.
Conjugation with antibody fragments or Fc domains may extend half-life, while fusion with targeting moieties
(e.g., chemokine receptors, integrins) could direct cytokine activity to inflamed tissues.
Beyond protein modifications, novel delivery platforms using programmable RNA or DNA vectors encoding the IL-2 sequence present a transformative approach. These systems could enable endogenous, in vivo production of the cytokine under the regulation of tunable promoters, offering a pathway for sustained yet reversible immune modulation. The ultimate integration of these sophisticated molecular tools with biocompatible delivery systems and real-time immune monitoring is poised to give rise to a new class of "smart cytokines," capable of dynamically adapting their activity to the evolving immunological state of the patient.
Conclusion
Low-dose IL-2 has transformed from a conventional cytokine into precision immunomodulation, demonstrating that immune responses can be selectively and restoratively recalibrated rather than broadly suppressed. The coming decade will likely see this approach mature into a mainstream clinical modality. Its sustained success will rest on three critical pillars: a deepening mechanistic understanding of Treg biology, relentless technological innovation in cytokine design and delivery, and the implementation of data-driven personalization strategies. By systematically addressing the remaining challenges including stability, variability, and precise tissue targeting, Ld-IL-2 has the potential to evolve from a promising biological therapy into a cornerstone of immune-tolerance medicine, reshaping therapeutic strategies across autoimmunity, transplantation, and chronic inflammatory diseases.
Abbreviations
Tregs: Regulatory T cells; pTregs: peripherally induced Tregs; TR-Tregs: Tissue-resident Tregs; Ld-IL-2: low-dose interleukin-2; NK cell: natural killer cell; Th17: T helper 17; Tfh: T follicular helper; Teffs: effector T cells; SLE: Systemic lupus erythematosus; Bcl-2: B cell lymphoma 2; TSDR: Treg-specific demethylated region; DNMT: DNA methyltransferase; OXPHOS: oxidative phosphorylation; AMPK: AMP-activated protein kinase; ROS: reactive-oxygen-species; SAM: S-adenosylmethionine; EVs: extracellular vesicles; IFN-γ: interferon-gamma; APC: antigen-presenting cell; RP: Relapsing polychondritis; CRP: C-reactive protein; RPDAI: Relapsing Polychondritis Disease Activity Index; PLA: polylactic-acid; HDAC: histone deacetylase.
Declarations
Author contributions
Ruiling Feng: writing original draft, prepare, create, or express the content for publication, especially in writing the initial draft. Bo Huang and Xia Zhang: writing review and editing, prepare, create, or express the content for publication, especially in writing the initial draft. Jing He: supervision, supervise and lead the planning and execution of research activities. All authors read and approved the final manuscript.
Acknowledgements
Not Applicable.
Funding information
This study was funded by National Key Research and Development Program of China (2022YFE0131700), National Natural Science Foundation of China (82271835, 82071813).
Ethics approval and consent to participate
Not Applicable.
Competing Interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from the authors.
References
 Figures
Figures References
References Peer
Peer Information
InformationFigure 1. Mechanistic network of low-dose IL-2 in immune regulation. Low-dose IL-2 preferentially signals through the high-affinity IL-2 receptor (IL-2Rαβγ) on regulatory T cells, engaging JAK–STAT and mTOR–AMPK signaling. These pathways sustain Foxp3 transcription and reprogram cellular metabolism, thereby stabilizing Treg identity and function. The resulting network suppresses Th17 and Tfh responses while maintaining CD8+ T and NK cell homeostasis, collectively restoring immune balance.
Figure 2. Dose-dependent biphasic effects of IL-2 on Treg cells. Low-dose IL-2 induces a selective, dose-dependent expansion of CD4+CD25hiFoxp3+ Tregs through engagement of the high-affinity IL-2Rαβγ complex, with maximal response at approximately 1×10⁶ IU. Higher doses may activate conventional T (Tconv) and NK cells, thereby diminishing Treg selectivity and shifting the immune balance toward activation.
Figure 3. Engineered IL-2 variants and targeted delivery strategies. (A) Topological Engineered IL2 Variants. Rational IL2 mutation to reduce IL-2Rβ affinity while preserving IL-2Rα binding, achieving CD25-biased agonists with enhanced Treg selectivity, prolonged half-life, and higher stability. (B) Targeted Delivery for Sustained Release. A hybrid nanoparticle platform combining PLA microspheres and exosomes enables designed for controlled IL-2 release, maintaining therapeutic concentrations within the selective window. (C) Tissue-Selective Immunomodulation. Local injection including salivary, gland and joint concentrates IL-2 at sites of inflammation, maximizing Treg enrichment and resulting in reduced inflammation with limited systemic toxicity.
Peer-review Terminology
Identity transparency: Single anonymized
Reviewer interacts with: Editor
Details
This is an open access article under the terms of the Creative Commons Attribution License(http://creativecommons.org/licenses/by/4.0/), which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Publication History
Received 2025-10-23
Accepted 2026-01-15
Published 2026-03-10