




Abstract
Background:Myasthenia Gravis (MG) is a chronic autoimmune neuromuscular disorder that severely impacts patients' quality of life. Identifying plasma and cerebrospinal fluid (CSF) proteins with a genetic causal relationship to MG may provide novel therapeutic targets.
Methods:This study employed the Mendelian randomization (MR) approach, in combination with Bayesian colocalization analysis, to assess the causal relationship between 4,185 plasma proteins and 832 CSF proteins and the risk of MG. Sensitivity analyses were also performed to validate the robustness of the MR results. Additionally, protein-protein interaction networks and candidate drug predictions were utilized to elucidate the complex interactions between proteins and identify potential drug targets.
Results:Three plasma proteins and five CSF proteins were significantly associated with MG risk. ALDH2, HSPA1A, PRSS8, MFRP, CTSH, SHBG, and TXNDC12 were found to increase MG risk, while IL36A was negatively correlated. Further colocalization analysis revealed strong evidence for the associations between PRSS8 and HSPA1A with MG (pph4 > 0.8), and substantial evidence for TXNDC12 and ALDH2 (0.8 > pph4 > 0.6).
Conclusion:This study employed proteomics-based MR to identify several plasma and CSF proteins significantly associated with the risk of MG. Notably, PRSS8, HSPA1A, TXNDC12, and ALDH2 emerge as potential therapeutic targets for MG. While these findings offer valuable insights into the pathological mechanisms of MG and the development of novel therapeutic strategies, further research is required to evaluate the feasibility and clinical efficacy of these candidate proteins.
Keywords: Myasthenia Gravis, Mendelian Randomization, Proteomics, Cerebrospinal Fluid Proteins, Colocalization, Therapeutic Target
Introduction
Myasthenia Gravis (MG) is a chronic autoimmune neuromuscular junction disorder [1]. Ocular weakness is the most common initial symptom, but the condition often progresses to involve the medullary muscles, limbs, axial muscles, and respiratory muscles, ultimately leading to generalized MG [2]. Studies indicate that the global prevalence of MG is approximately 20 to 50 cases per 100,000 people, with an annual incidence ranging from 0.3 to 2.8 cases per 100,000 people. MG can occur at any age, though it is most common in young women and elderly men [3]. Standard treatment options include acetylcholinesterase inhibitors, corticosteroids, and immunosuppressants [4]. For patients who show inadequate responses to drug therapy, thymectomy, intravenous immunoglobulin, and plasmapheresis are commonly utilized [5]. Although these treatments help control symptoms, they still have significant limitations. Drug therapy often requires lifelong treatment, which potentially lead to side effects such as gastrointestinal discomfort, muscle spasms, and an increased risk of infections [5, 6]. Plasmapheresis and immunoglobulin therapy provide rapid symptom relief but have a short duration of effect and are costly [7]. Thymectomy may benefit some patients, but it carries surgical risks, and its effectiveness in cases of late-onset or antibody-negative MG remains uncertain [5, 8]. Additionally, approximately 10-20% of patients show limited or no response to standard treatments [5], highlighting the urgent need for further research into the pathological mechanisms of MG and the identification of novel therapeutic targets.
Proteins play a crucial role in the pathophysiology of MG. In MG patients, autoantibodies bind to target antigens at the neuromuscular junction, activating the complement system, inflammation, and the damage or functional alteration of acetylcholine receptors, thus impairing neuronal signal transmission [9]. The circulating proteome comprises proteins from various cells and tissues throughout the body, which may be actively secreted into the bloodstream or passively released during cell damage or turnover, serving as biomarkers for disease [10]. A cohort study of MG patients found elevated serum amyloid A (SAA) levels in MG patients and promoted the expansion of CD4+ T cells and CD19+ B cell subpopulations [11]. Furthermore, IL-17 expression levels were correlated with the severity of the disease in MG patients, suggesting its potential promotive role in the pathogenesis of MG [12]. While no direct studies have yet utilized cerebrospinal fluid (CSF) proteins to identify therapeutic targets for MG, a review article pointed out the importance of CSF analysis in MG patients to better understand the immune pathological processes associated with the disease, particularly for developing personalized treatment strategies for different MG subtypes [13]. Moreover, proteins are the targets of most pharmacological agents [14]. A randomized, double-blind, placebo-controlled, multicenter phase 3 clinical trial (ADAPT study, NCT03669588) demonstrated that efgartigimod improved clinical symptoms in MG patients by inhibiting the neonatal Fc receptor, significantly enhancing their quality of life [15]. However, despite the identification of associations between certain circulating proteins and MG, clarifying their causal relationship is hindered by factors such as small sample sizes and observational study designs. Conducting large-scale randomized controlled trials to explore potential causal links between numerous proteins and MG remains impractical.
Mendelian Randomization (MR) is a method that uses genetic variation as an instrumental variable to assess causal relationships between exposures and outcomes. Genetic variations are determined before birth and are generally not influenced by postnatal environmental or behavioral factors, effectively minimizing the impact of confounding variables [16]. Proteomics-based MR, which integrates pQTL of plasma and CSF proteins and genome-wide association study (GWAS) data on MG, analyzes proteins that may influence MG, thereby completely avoiding reverse causality. As such, proteomics-based MR offers a novel approach to elucidating the molecular mechanisms underlying MG and can also aid in identifying genetically supported drug therapy targets. This approach has the potential to enhance the success rate of clinical drug development.
Methods
Study Design
In this study, we utilized pQTL data from large-scale plasma and CSF proteomics studies and
applied MR to investigate the genetic causal relationship between these proteins and MG. Additionally, we conducted
protein-protein interaction (PPI) network construction and colocalization analysis on the statistically significant
proteins identified in the MR analysis, with the goal of pinpointing the most promising therapeutic targets and
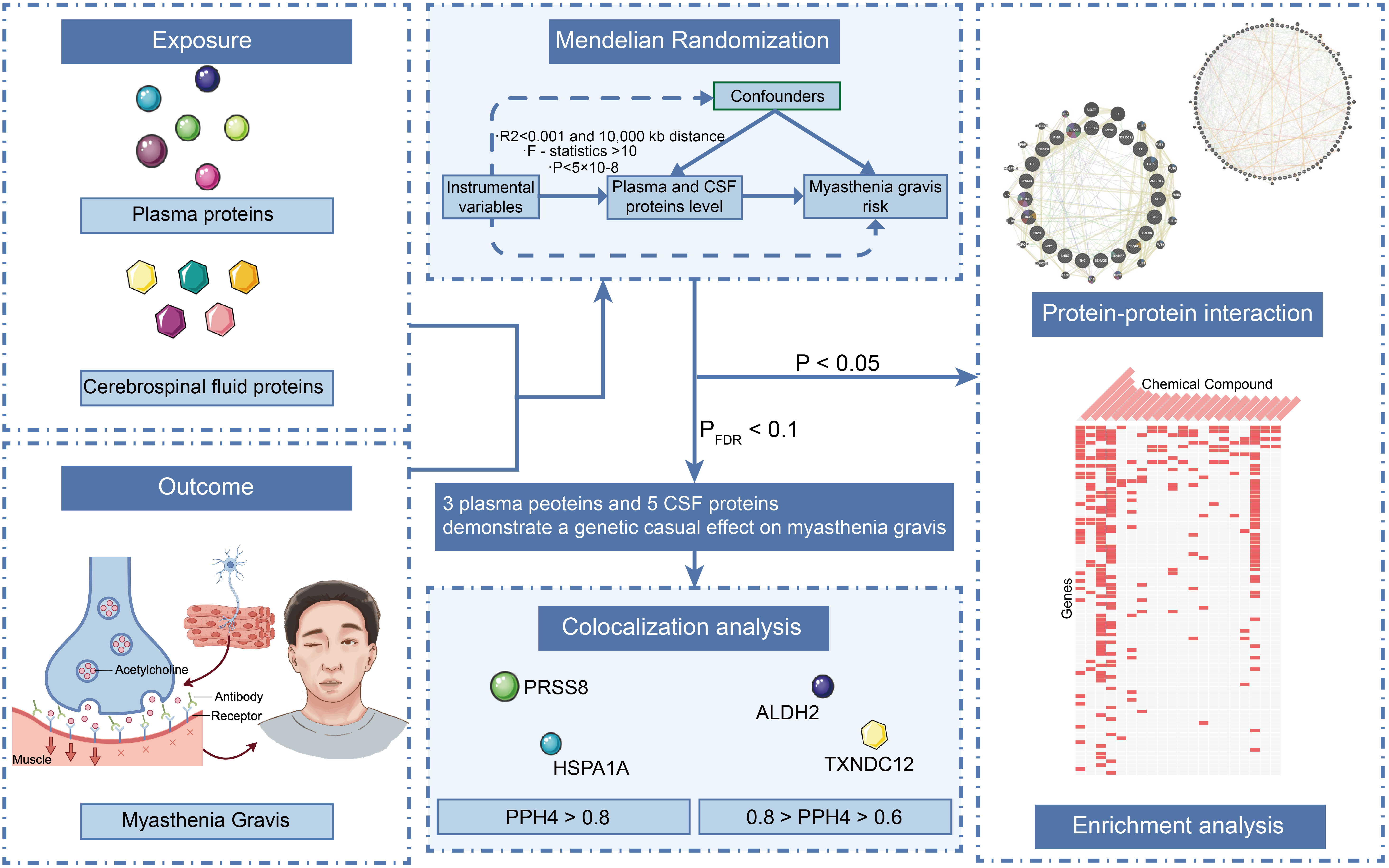
predicting potential drugs. This approach aims to bridge basic research and clinical applications (Figure 1).
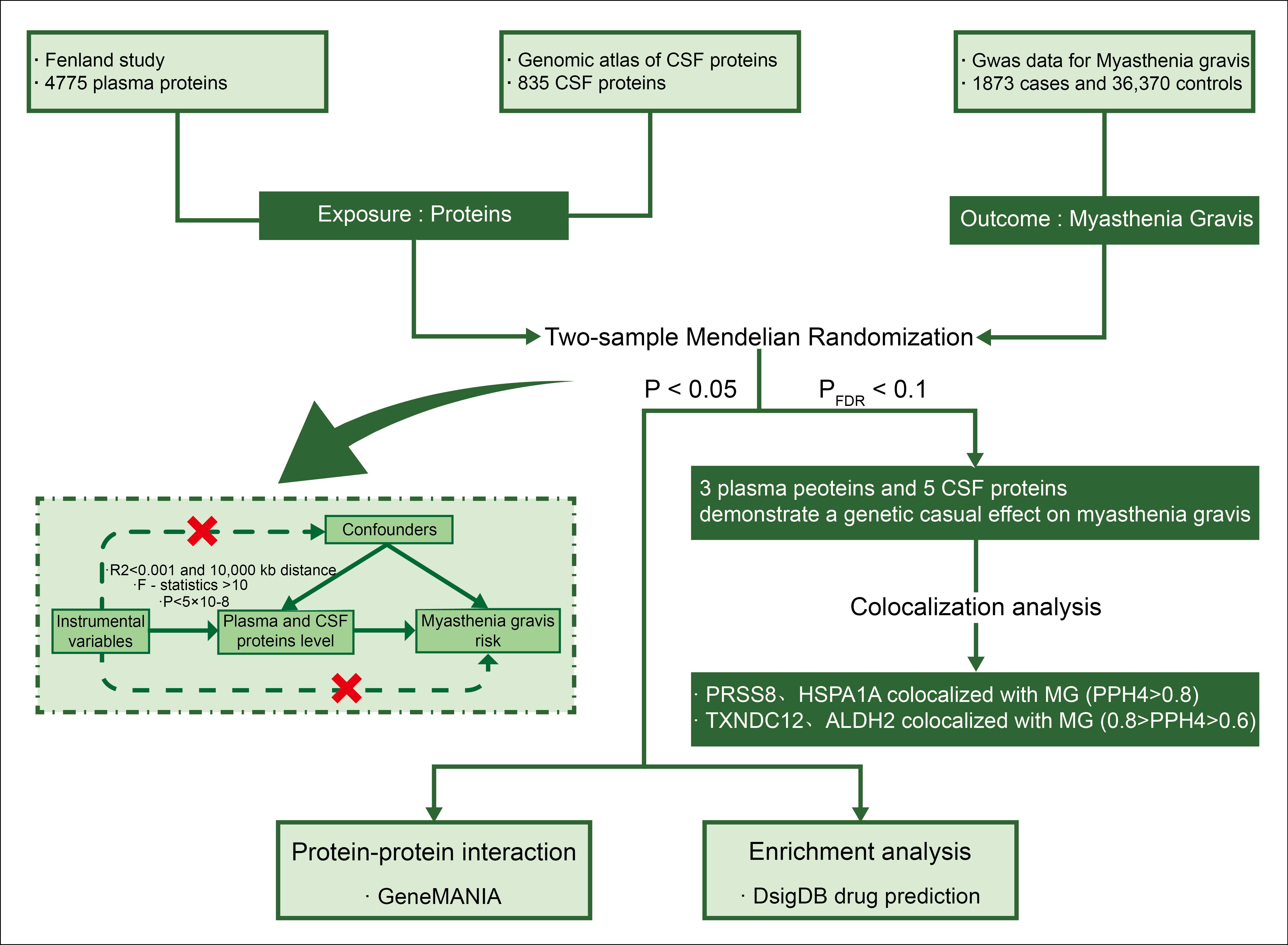
Figure 1. Flow Chart of the Overall Study Design. It begins with the exposure of proteins derived from the Fenland study (4775 plasma proteins) and the Genomic Atlas of CSF proteins (835 proteins). The outcome is MG, based on GWAS data for 1873 cases and 36,370 controls. The study employs two-sample Mendelian randomization to assess causal relationships, with significant findings (P < 0.05 and PFDR < 0.1) showing that three plasma proteins and five CSF proteins have a genetic causal effect on MG. Further analysis includes colocalization and enrichment analyses.
Exposure Data Source
We obtained single nucleotide polymorphisms (SNPs) data associated with plasma
protein levels from the Fenland study. This genome-proteome association study included 10,708 participants of European
descent and collected data on 4,775 plasma proteins using SomaScan v4 (http://www.omicscience.org/apps/pgwas) [17]. Since the selected circulating proteins were based on cis-acting protein quantitative trait loci (cis-pQTL), a
total of 4,185 plasma proteins were included in the subsequent analysis.
The SNP data associated with CSF protein levels were derived from a genome-proteome association study conducted by the Washington University School of Medicine, which included 971 participants. Using an aptamer-based high-throughput proteomics platform, 1,305 proteins were detected. Following stringent quality control, 835 CSF protein data points were obtained [18]. After excluding duplicate-sequenced proteins, 832 CSF proteins were included in the subsequent analysis.
Outcome Data Source
In this study, our outcome data were sourced from the largest MG meta-GWAS study
conducted in the United States and Italy. The study included 1,873 acetylcholine receptor antibody-positive (AChR+) MG
patients and 36,370 healthy controls, excluding all muscle-specific kinase antibody-positive (MuSK+) patients. Data
were collected through collaboration among institutions including Johns Hopkins University, the National Institute on
Aging, the University of Pisa, and the Catholic University of Rome, with ethical approval from all participating
institutional review boards [19].
Selection of Instrumental Variables
In conducting the MR analysis, we selected cis-pQTLs that directly
regulate protein expression levels as instrumental variables (IVs), with cis-pQTLs spanning a gene range of ±1Mb. The
selection of SNPs was performed based on three main assumptions: 1) SNPs highly correlated with plasma proteins were
selected, and these SNPs met the significance threshold (P-value < 5 × 10-8); 2) The linkage disequilibrium (LD)
parameter threshold (r2) was set to 0.001, and SNPs were required to be at least 10,000 kb apart to ensure
independence between SNPs and minimize the impact of LD; 3) Weak IVs, defined as those with an F-statistic < 10 were
excluded to ensure a stable and reliable association with the exposure; 4) SNPs strongly associated with the outcome
(P-value > 5 × 10-8) were excluded.
Statistical Analysis
In this MR study, we employed several statistical methods to assess the potential
causal relationship between circulating proteins and MG. When two or more IVs were available, the inverse variance
weighted (IVW) method was chosen as the primary analytical tool [20]. For proteins with only a
single instrumental variable, the Wald ratio method was applied to estimate the change in the log-odds ratio of MG
risk for each standard deviation (SD) increase in protein levels. To control for the expected false-discovery rate, we
applied the Benjamini-Hochberg (B-H) correction to adjust for p-values resulting from multiple tests and set the
statistical significance threshold at PFDR < 0.1 to enhance the reliability of the results [21].
Additionally, we conducted sensitivity analyses using Cochran's Q test, MR-Egger, and MR-PRESSO methods.Cochran's Q statistic was used to assess the heterogeneity among the selected IVs, with a P-value < 0.05 indicating significant heterogeneity. The MR-Egger method evaluates causality under the InSIDE assumption and can be performed even in the absence of valid IVs. A significant intercept in the MR-Egger method suggests the possibility of pleiotropy, indicated by a P-value < 0.05. The MR-PRESSO method, implemented through the "MR-PRESSO" package, identifies and removes SNP outliers with horizontal pleiotropy. However, in cases where the number of SNPs is small, it may be insufficient for effective analysis of heterogeneity and pleiotropy [22]. Finally, the MR-Steiger test assesses the directionality of causal effects by comparing the variance ratio of IVs between the exposure and outcome variables, thus evaluating the applicability of the IVs [23].
pQTL-GWAS Colocalization Analysis
To assess whether two traits share causal variants in a single region,
we performed colocalization analysis using the default prior probabilities of the coloc R package [24]. For each cis-protein gene locus, the Bayesian method tested five mutually exclusive hypotheses: 1) no association
with either trait (H0); 2) only associated with protein levels (H1); 3) only associated with MG risk (H2); 4)
associated with both protein levels and MG risk, but with independent genetic variants for each trait (H3); 5) protein
levels and MG risk share the same genetic variants (H4). In this study, the degree of colocalization support was
measured using the posterior probability (pph4).
PPI Network and Potential Drug Prediction
Due to the presence of the blood-brain barrier (BBB), the
connection between plasma pQTLs and CSF pQTLs may be relatively weak. To further identify protein targets associated
with MG risk and to understand protein interactions in different tissue environments, we constructed PPInetworks for
plasma and CSF proteins using the GeneMANIA tool [25].
Proteins are the fundamental units of bodily functions and represent one of the key categories of druggable targets [26], this study subsequently utilized the Drug Signatures Database (DSigDB) to predict potential drugs and assess the druggability of genes corresponding to these target proteins [27]. The database contains 22,527 gene sets, encompassing 17,389 compounds and 19,531 genes, offering researchers a powerful tool for identifying and validating potential drug-target genes.
Results
Plasma and CSF Proteins and MG Risk: MR Analysis
Our team employed the IVW method and the Wald ratio
method to comprehensively assess the effects of 4,185 plasma pQTLs and 832 CSF pQTLs on the risk of MG. Ultimately, we
identified 95 plasma proteins and 23 CSF proteins associated with MG risk (P < 0.05). We then applied B-H correction
to adjust the p-values, and the results indicated that three plasma proteins—Aldehyde Dehydrogenase 2 (ALDH2), Heat
Shock Protein Family A Member 1A (HSPA1A), and Serine Protease 8 (PRSS8)—and five CSF proteins—Interleukin 36 Alpha
(IL36A), Membrane Frizzled-Related Protein (MFRP), Cathepsin H (CTSH), Sex Hormone Binding Globulin (SHBG),
Thioredoxin Domain Containing 12 (TXNDC12)—have a genetic causal relationship with MG risk (PFDR < 0.1).
Specifically, higher expression levels of plasma ALDH2 (OR = 2.32, 95% CI: 1.48-3.62, P = 2.22e-04, PFDR =
0.079), HSPA1A (OR = 2.68, 95% CI: 1.59-4.51, P = 2.16e-04, PFDR = 0.079), and PRSS8 (OR = 6.89, 95% CI:
3.00-15.86, P = 5.62e-06, PFDR = 0.008) increase the risk of MG (Figure 2). Regarding CSF
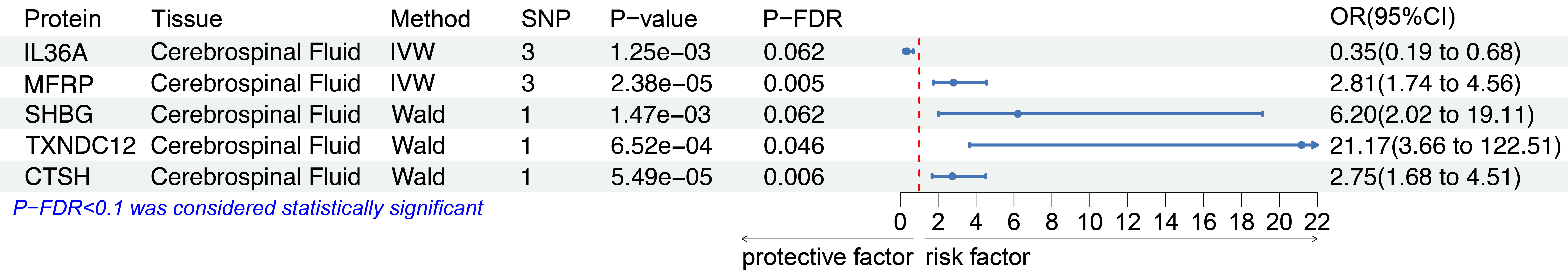
proteins, higher expression levels of MFRP (OR = 2.81, 95% CI: 1.74-4.54, P = 2.38e-05, PFDR = 0.005), CTSH
(OR = 2.75, 95% CI: 1.68-4.49, P = 5.49e-05, PFDR = 0.006), SHBG (OR = 6.20, 95% CI: 2.02-19.09, P =
1.47e-03, PFDR = 0.062), and TXNDC12 (OR = 21.17, 95% CI: 3.66-122.49, P = 6.52e-04, PFDR =
0.046) were found to increase the risk of MG, whereas higher expression of IL36A (OR = 0.35, 95% CI: 0.19-0.66, P =
1.25e-03, PFDR = 0.062) was negatively correlated with MG risk (Figure 3). Detailed data
are shown in the Supplementary Materials.
Figure 2. Forest Plot Displaying Plasma Proteins Genetically Associated with MG. The data shown represent the odds ratios with 95% confidence intervals, where PFDR < 0.1 was considered statistically significant. Proteins identified include PRSS8, HSPA1A, and ALDH2, with the corresponding P-values and PFDR values indicated for each protein.
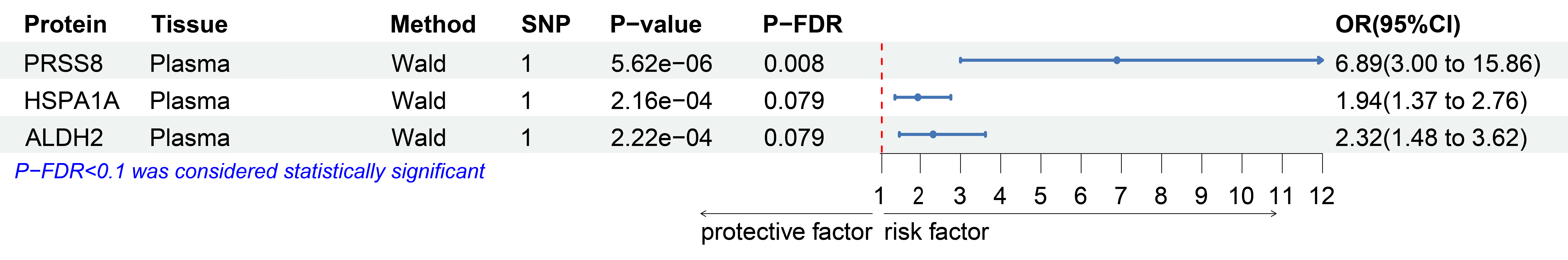
Figure 3. Forest Plot Displaying CSF Proteins Genetically Associated with MG. The data shown represent the odds ratios with 95% confidence intervals, where PFDR < 0.1 was considered statistically significant. Proteins identified include IL36A, MFRP, SHBG, TXNDC12, and CTSH, with the corresponding P-values and PFDR values indicated for each protein.
Sensitivity analysis further confirmed the reliability of the study findings. MR-Steiger testing validated the positive causal effects of the eight identified proteins on MG risk. Additionally, Cochran's Q test, MR-Egger, and MR-PRESSO analysis for proteins with more than three SNPs all yielded p-values greater than 0.05, indicating no significant heterogeneity or horizontal pleiotropy among the selected IVs.
Bayesian Colocalization Analysis
The Bayesian colocalization results (Table 1) revealed that PRSS8 and
HSPA1A exhibited strong colocalization with MG (pph4 > 0.8), suggesting these proteins share the same genetic variants
as MG and can be considered as primary drug candidate targets [28]. Meanwhile, TXNDC12 and ALDH2
showed pph4 values between 0.6 and 0.8, indicating moderate colocalization strength, and may be considered as
secondary candidate therapeutic targets. Detailed data are shown in the Supplementary Materials.
Table 1. The pph4 values from the colocalization analysis of proteins with \( P_{\text{pos}} < 0.1 \).
| Protein | pph4 |
|---|---|
| PRSS8 | 9.85E-01 |
| HSPA1A | 8.82E-01 |
| TXNDC12 | 7.31E-01 |
| ALDH2 | 6.51E-01 |
| SHBG | 3.96E-01 |
| CTSH | 1.26E-02 |
| IL36A | 1.62E-05 |
| MFRP | 2.53E-06 |
PPI Network and Potential Drug Prediction
Plasma and CSF proteins significantly associated with MG risk (P
< 0.05) were input into GeneMANIA for protein network construction. In addition to the input proteins, each network
was expanded to include 20 potential interacting genes. The plasma protein network was primarily driven by
co-expression mechanisms (55.75%), predictive models (17.10%), physical interactions (11.86%), and colocalization
(9.29%). The functions of this network mainly included axonogenesis, glycosaminoglycan binding, neuronal projection
guidance, regulation of chemotaxis, positive regulation of cell adhesion, ERK1 and ERK2 signaling cascades, and
positive regulation of α-β T cell activation (PFDR < 0.05). Detailed data are shown in the Supplementary Materials.
The CSF protein network was primarily connected through co-expression mechanisms (63.96%), with additional connections involving colocalization (16.67%), shared protein domains (14.07%), and genetic interactions (5.30%). The network's functions mainly include fucosyltransferase activity, positive regulation of cell adhesion, CD4-positive α-β T cell cytokine production, and T cell- and lymphocyte-mediated immunity, which form the core mechanisms of the immune response. Detailed data are shown in the Supplementary Materials. Notably, IL18 was identified as a potential interacting gene in both protein networks.
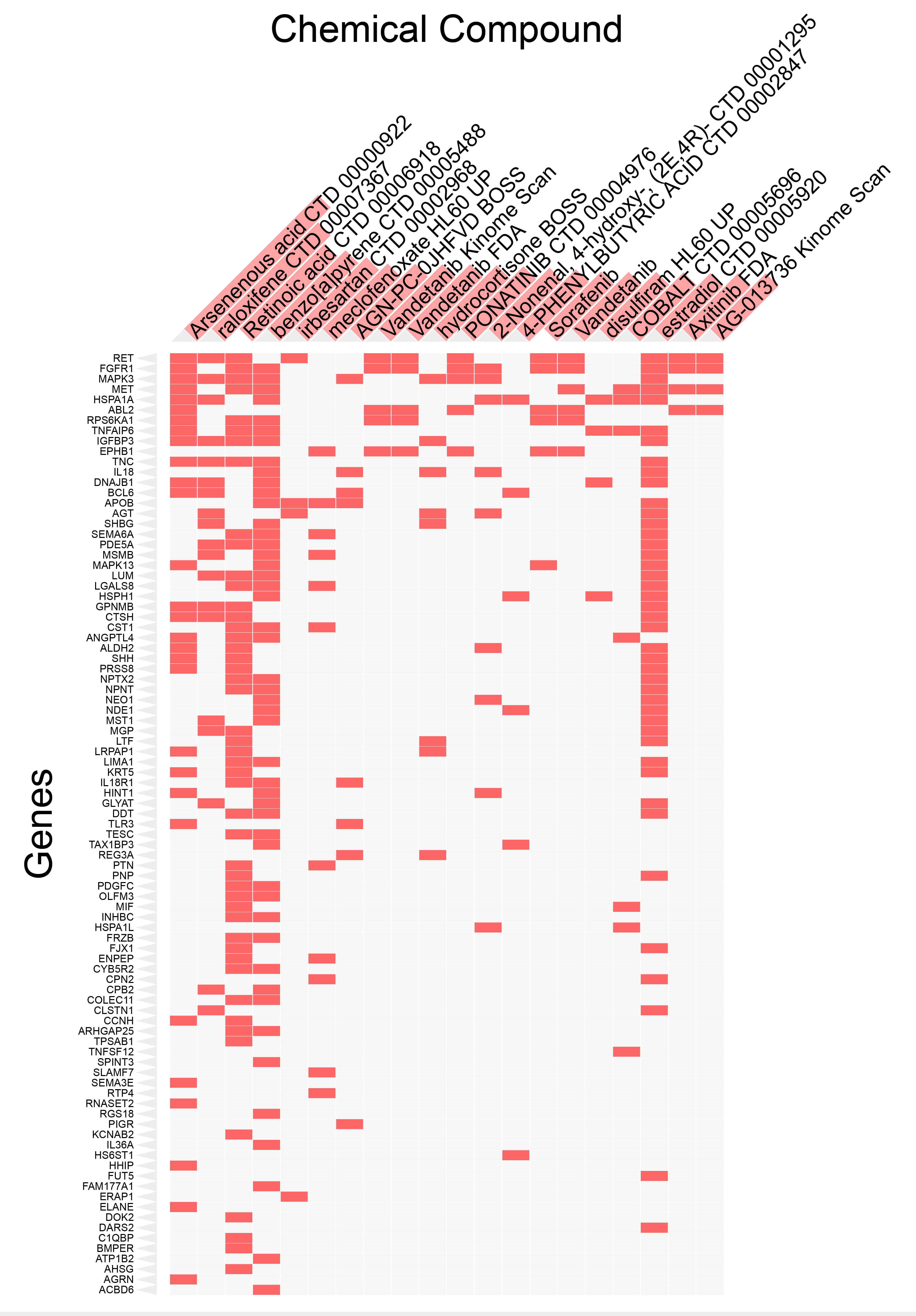
Subsequently, we compiled the same-named genes of IL18 and the significant proteins from the two proteomics datasets and used the DSigDB database via the open-source Enrichr platform (https://maayanlab.cloud/Enrichr/) to predict candidate drugs associated with these genes [29]. The results were ranked by adjusted p-values, from smallest to largest. Figure 4 displays the clustering of the top 20 chemical compounds with adjusted p-values less than 0.05, along with all target genes. The results show that arsenous acid (Arsenous acid CTD 00000922) had the smallest adjusted p-value, indicating the highest significance. Most of the genes interacted with retinoic acid (Retinoic acid CTD 00006918), benzo(a)pyrene (benzo[a]pyrene CTD 00005488), and estradiol (estradiol CTD 00005920). Detailed data are shown in the Supplementary Materials.
Figure 4. Top 20 Chemical Compounds Targeting Genes Corresponding to Proteins Associated with MG Risk. This figure visualizes the results from the DSigDB database via the Enrichr platform, which predicts candidate drugs associated with genes whose corresponding proteins are linked to MG risk (P < 0.05). The chemical compounds are ranked by adjusted p-values, with the top 20 compounds having p-values less than 0.05 displayed. The red bars along the horizontal axis represent the strength of the association between each chemical compound and the corresponding genes, with longer bars indicating stronger associations.
Discussion
To our knowledge, this study is the first to combine plasma and CSF proteomics to explore new therapeutic targets for MG. Through rigorous multi-condition restrictions, we identified three plasma proteins and five CSF proteins that have a significant genetic causal relationship with MG risk. The results demonstrate that high expression of PRSS8, HSPA1A, TXNDC12, ALDH2, SHBG, CTSH, MFRP, and low expression of IL36A are positively associated with MG susceptibility. Among these, PRSS8 and HSPA1A showed strong colocalization support (pph4 > 0.8), while TXNDC12 and ALDH2 exhibited moderate colocalization support (0.8 > pph4 > 0.6). Additionally, the complex interactions between proteins were visualized through the PPInetwork. Based on the expanded results from both plasma and CSF protein interaction networks, we inferred that IL18 is also closely related to MG risk. Finally, drug predictions may facilitate the translation of basic research findings into clinical applications, optimize the research and development process, and reduce drug development costs.
PRSS8, also known as Prostasin, is a member of the serine protease family. Previous studies have shown that PRSS8 (Prostasin) is co-expressed with Matriptase during embryonic development, and that Prostasin can indirectly participate in extracellular matrix remodeling by activating Matriptase [30]. Additionally, Prostasin influences the migration and adhesion of immune cells, a mechanism confirmed in the tumor microenvironment [31]. We hypothesize that, during the pathogenesis of MG, PRSS8 may contribute to abnormal attacks on the neuromuscular junction by affecting the migration and localization of lymphocytes, thereby exacerbating disease progression. Moreover, one study suggests that PRSS8 can regulate the Toll-like receptor 4 (TLR4)-mediated signaling pathway, affecting the expression of inflammatory factors such as TNF-α, IL-6, and IL-1β [32]. Another study indicates that PRSS8 can modulate the expression of inflammatory factors (including IL-6 and IL-8) in prostate epithelial cells through the regulation of PAR-2 and associated signaling pathways [33]. These inflammatory factors, including TNF-α, IL-6, and IL-1β, have been established as key regulators in the autoimmune response of MG [34]. Based on existing research and our MR analysis, we propose that changes in the expression levels and activity of PRSS8 could be a crucial molecular event leading to the onset of MG. Future studies should focus on investigating the expression of PRSS8 and its related signaling pathways in MG patients to further validate this hypothesis.
HSPA1A, a member of the heat shock protein 70 (HSP70) family, primarily functions as a molecular chaperone that prevents the aggregation of misfolded proteins [35]. It plays a central role in various biological processes, including stress response, signal transduction, and cell cycle regulation [36]. HSPA1A can bind to peptide fragments generated within the cell due to stress or injury and subsequently release them extracellularly. These HSPA1A-peptide complexes can be recognized and internalized by antigen-presenting cells (APCs), where they are presented to T cells via MHC molecules, subsequently triggering a specific T cell response against self-antigens [37]. In the context of MG, if these peptides originate from the acetylcholine receptor, they may lead to misrecognition of these self-antigens by T cells, activating B cells to produce corresponding autoantibodies, thus exacerbating the disease. Furthermore, the interaction between HSPs and APCs can promote the secretion of inflammatory cytokines by macrophages, further amplifying the autoimmune response [37, 38]. Interestingly, studies have shown that compared to healthy controls, the mRNA expression level of HSPA1A in the peripheral blood mononuclear cells (PBMCs) of MG patients is significantly reduced [35]. This suggests a more complex potential relationship than initially expected, warranting further research to resolve this paradox and better understand the role of HSPA1A in the pathogenesis of MG [35].
TXNDC12, also known as AGR1, TLP19, or ERP18/19, is a member of the protein disulfide isomerase (PDI) family and plays a crucial role in the proper folding of proteins [39]. While research on the relationship between TXNDC12 and the risk of MG remains limited, an increasing body of evidence suggests that sustained protein misfolding can initiate apoptotic cascades, contributing to the development of various neurological diseases [40]. ALDH2 is primarily responsible for the metabolism of acetaldehyde in the body and is a key enzyme in maintaining cellular metabolic balance and reducing oxidative stress [41]. Like TXNDC12, the association between ALDH2 and MG risk has been infrequently reported. However, in this multi-omics MR study, the levels of TXNDC12 and ALDH2 were found to exhibit strong positive correlations with MG risk. This genetic finding implies a complex pathophysiological relationship between TXNDC12, ALDH2, and MG. Therefore, future research should focus on elucidating the biological functions of these proteins and the associated molecular pathways involved in the pathogenesis of MG, with the aim of identifying potential new therapeutic targets for the disease.
This study leveraged a large-scale Finnish plasma protein dataset, along with data from over 900 CSF proteins, and the largest available GWAS on MG to conduct a MR analysis, thereby enhancing the robustness of the findings. Additionally, we employed multiple methodological strategies to minimize potential confounding factors. Specifically, we used the Steiger test to mitigate the influence of reverse causality. For plasma proteins, we prioritized cis-pQTLs, located at or near the gene encoding the target proteins, over trans-pQTLs and eQTLs due to their substantial contribution to explaining protein expression [42]. The B-H correction was applied to control for the false-positive rate, and gene colocalization analysis was conducted to further enhance the robustness of the statistical results. Finally, through PPInetworks and potential drug predictions, we offer novel insights for the development of MG therapies.
However, our study has several limitations. First, this research is based on data from populations of European ancestry, which limits the generalizability of the results to different ancestral groups. Second, although the study encompassed a broad range of proteins, it may have overlooked other potential therapeutic targets due to the selection of variables based on stringent significance thresholds. Furthermore, drug predictions and the construction of interaction networks were based on the gene names corresponding to the proteins. The function and regulation of proteins are influenced by multiple factors, including environmental interactions and epigenetic modifications, which were not fully considered in this study, potentially oversimplifying the pathways through which proteins affect MG. Finally, MG exists in two subtypes, AChR+ and MuSK+, but we did not distinguish between these subtypes when selecting the outcome data sources, potentially overlooking target specificity.
Conclusion
In conclusion, our study suggests that the levels of three plasma proteins and five CSF proteins are causally associated with the risk of MG. Among these, PRSS8, HSPA1A, TXNDC12, and ALDH2 are promising candidates for new therapeutic targets. Our research provides new perspectives for understanding the pathogenesis of MG. However, further studies are needed to confirm the association between these candidate plasma proteins and MG risk to establish their clinical relevance.
Abbreviations
Acetylcholine receptor antibody-positive: AChR+; Aldehyde De-hydrogenase 2: ALDH2; Benjamini-Hochberg: B-H; Blood-brain barrier: BBB; Cathepsin H: CTSH; Cerebrospinal fluid: CSF; Cis-acting protein quantitative trait loci: cis-pQTL; Drug Signa-tures Database: DSigDB; Heat Shock Protein Family A Member 1A: HSPA1A; Heat shock protein 70: HSP70; Instrumental variables: IVs; Interleukin 36 Alpha: IL36A; Inverse variance weighted: IVW; Membrane Frizzled-Related Protein: MFRP; Mendelian Randomization: MR; Myasthenia Gravis: MG; Mus-cle-specific kinase antibody-positive: MuSK+; Peripheral blood mononuclear cells: PBMCs; Protein disulfide isomerase: PDI; Protein-protein interaction: PPI; Serine Protease 8: PRSS8; Se-rum amyloid A: SAA; Single nucleotide polymorphisms: SNPs; Standard deviation: SD; Toll-like receptor 4: TLR4.
Declarations
Author contributions
Hongmei Miao: Writing-review & editing, Writing-original draft, Conceptualization, Visualization, Formal analysis. Yinying Chai: Writing-review & editing, Validation, Writing-original draft, Supervision, Data curation, Formal analysis. Qianran Hong: Writing-review & editing, Validation, Formal analysis. Yuxuan Song: Writing-review & editing, Validation, Funding acquisition, Data curation. Yibo He: Writing-review & editing, Conceptual-ization, Supervision. All authors read and approved the final manuscript.
Acknowledgements
Not Applicable
Funding information
This work was supported by Innovation Fund for Qutstanding Doctoral Candidates of Peking University Health Science Cen-ter (BMU2024BSS001).
Ethics approval and consent to participate
Not Applicable.
Competing Interests
The authors declare that they have no existing or potential commercial or financial relationships that could create a con-flict of interest at the time of conducting this study.
Data availability
The data that supports the findings of this study are available in the supplementary materials of this article.
References
 Figures
Figures References
References Peer
Peer Information
InformationFigure 1. Flow Chart of the Overall Study Design. It begins with the exposure of proteins derived from the Fenland study (4775 plasma proteins) and the Genomic Atlas of CSF proteins (835 proteins). The outcome is MG, based on GWAS data for 1873 cases and 36,370 controls. The study employs two-sample Mendelian randomization to assess causal relationships, with significant findings (P < 0.05 and PFDR < 0.1) showing that three plasma proteins and five CSF proteins have a genetic causal effect on MG. Further analysis includes colocalization and enrichment analyses.
Figure 2. Forest Plot Displaying Plasma Proteins Genetically Associated with MG. The data shown represent the odds ratios with 95% confidence intervals, where PFDR < 0.1 was considered statistically significant. Proteins identified include PRSS8, HSPA1A, and ALDH2, with the corresponding P-values and PFDR values indicated for each protein.
Figure 3. Forest Plot Displaying CSF Proteins Genetically Associated with MG. The data shown represent the odds ratios with 95% confidence intervals, where PFDR < 0.1 was considered statistically significant. Proteins identified include IL36A, MFRP, SHBG, TXNDC12, and CTSH, with the corresponding P-values and PFDR values indicated for each protein.
Figure 4. Top 20 Chemical Compounds Targeting Genes Corresponding to Proteins Associated with MG Risk. This figure visualizes the results from the DSigDB database via the Enrichr platform, which predicts candidate drugs associated with genes whose corresponding proteins are linked to MG risk (P < 0.05). The chemical compounds are ranked by adjusted p-values, with the top 20 compounds having p-values less than 0.05 displayed. The red bars along the horizontal axis represent the strength of the association between each chemical compound and the corresponding genes, with longer bars indicating stronger associations.
Peer-review Terminology
Identity transparency: Single anonymized
Reviewer interacts with: Editor
Details
This is an open access article under the terms of the Creative Commons Attribution License(http://creativecommons.org/licenses/by/4.0/), which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Publication History
Received 2025-06-19
Accepted 2025-07-14
Published 2025-10-01